近視的發生發展與鞏膜重塑密切相關。因此,為了有效防治近視,闡明鞏膜重塑的發生機制至關重要。近年來,我國學者發現內質網應激可以通過未折疊蛋白反應中的肌醇需求蛋白-1/X盒結合蛋白-1通路調節凋亡蛋白的表達,從而參與調節缺氧狀態下鞏膜成纖維細胞的狀態調節鞏膜重塑的發生發展。與此同時,部分研究發現,通過藥物及遺傳學方法同時敲除內質網應激中的蛋白激酶RNA樣內質網激酶和轉錄激活因子6,可以有效抑制眼軸的增長。這證明了內質網應激在鞏膜重塑的發生發展過程中起到了重要作用。但對于內質網應激與鞏膜重塑的綜合分析國內外尚無報道。深入分析內質網與鞏膜重塑的關聯,對后續鞏膜重塑機制的分析研究具有重要意義。
近年來,近視的患病率不斷上升,已成為全球的公共衛生問題[1-2]。導致近視的主要原因是眼軸長度的改變,這一過程是鞏膜重塑的結果。一般認為,近視的發生發展與鞏膜重塑密切相關,鞏膜重塑是一個動態過程,其涉及了鞏膜細胞外基質(ECM)的合成與降解[3]。為了有效干預近視的進一步發生發展,分析及闡明鞏膜重塑的具體機制至關重要。已有研究證明,內質網應激(ERS)在維持細胞穩態和組織穩定性、纖維化疾病ECM重塑的發生發展以及近視相關鞏膜重塑中發揮重要作用[4-6]。但對于ERS與ECM重塑的綜合分析國內外尚無報道。現就ERS在鞏膜重塑中的研究現狀及進展作一綜述,以期為后續鞏膜重塑機制的分析研究提供參考。
1 鞏膜重塑
鞏膜是致密的纖維粘彈性結締組織,對維持眼球的形狀起著關鍵作用。其主要結構成分是由鞏膜成纖維細胞分泌的膠原纖維,其中Ⅰ型膠原蛋白構成的膠原纖維占比最高[7]。除此之外,Ⅰ型膠原蛋白亦是鞏膜ECM的主要組成成分[3]。鞏膜的胚胎發育伴隨著鞏膜成纖維細胞的生長和繁殖、膠原等的分泌、膠原類型和含量的變化以及膠原纖維的形成等[8]。膠原蛋白形成膠原纖維,使鞏膜具有很強的彈性,并保持鞏膜結構和眼球的形狀[9]。因此,膠原蛋白是研究鞏膜重塑的關鍵蛋白。在近視發生過程中,鞏膜是靶組織,而膠原蛋白則是所有調節機制的靶蛋白。近視發生的早期,鞏膜的Ⅰ型膠原蛋白mRNA水平降低,導致Ⅰ型膠原蛋白減少、鞏膜ECM成分改變,從而引起鞏膜變薄、眼軸增長及眼球的形態改變[10-11]。因此,鞏膜重塑的實質其實是鞏膜ECM的重塑,主要包括ECM mRNA表達和鞏膜生物力學性質的變化[12]。其中,膠原纖維的直徑、分布和方向都會影響鞏膜的生物力學特性。
目前,大量研究表明,鞏膜重塑的發生與脈絡膜變薄、血流減少密切相關[13]。脈絡膜是鞏膜血液供應的主要來源,其血流量的減少可能導致鞏膜缺氧,采用單細胞測序分析技術也發現近視眼的鞏膜中缺氧信號富集,由此證明近視的發生發展過程中鞏膜確實存在著缺氧現象[14]。
內質網是調控細胞內蛋白質正確折疊的重要細胞器,只有適當折疊的蛋白質才能被運送到高爾基體進行進一步加工[15]。當出現蛋白質過載、折疊能力減弱或者病理性刺激如缺氧時則可能誘發ERS并激活未折疊蛋白反應(UPR)[16]。目前,已有研究表明,ERS可以在轉錄及翻譯水平上調基質金屬蛋白酶(MMP)-2的表達[17]。這與既往研究發現的缺氧可以導致MMP-2上調以及形覺剝奪性近視(FDM)小鼠模型中MMP-2表達明顯升高相一致[18]。MMP-2可以降解鞏膜Ⅰ型膠原蛋白,導致膠原變薄、ECM成分改變從而導致鞏膜重塑并引起近視的發生[18]。這說明ERS在鞏膜重塑的發生中占有重要地位。
2 ERS
內質網是真核細胞中最大的細胞器之一,其在細胞中三分之一以上蛋白質的合成、折疊和結構成熟過程中均起著重要作用[19]。當蛋白質負荷和折疊能力之間的不平衡、營養缺乏以及缺氧等刺激因素出現時,作為回應,細胞激活了ERS并表現為一種稱為UPR的適應性機制[20]。UPR通過抑制蛋白質翻譯、誘導內質網相關分子伴侶促進未折疊蛋白質的折疊、激活內質網相關蛋白降解系統去除未折疊蛋白質以及促進細胞存活來恢復蛋白質的動態平衡。
UPR有三個分支,由位于內質網膜上的不同的ERS傳感器啟動,其分別是蛋白激酶RNA (PKR)樣內質網激酶(PERK)、肌醇需求酶-1(IRE1)以及轉錄激活因子-6(ATF6)。在生理情況下,這些蛋白質與ER伴侶蛋白即免疫球蛋白重鏈結合蛋白(BiP,也稱為GRP78)結合,并保持無活性狀態。當ERS發生時,BiP與上述三種傳感器蛋白解離,并觸發了UPR的三條分支[21]。UPR的三條分支均會通過蛋白質折疊需求和能力,重新調整到動態平衡,以便細胞能夠繼續生存和發揮功能[22]。然而,當長期慢性的ERS難以恢復內質網的正常功能時,UPR的三條分支將會通過激活CCAAT/增強子結合同源蛋白(CHOP,也稱為生長停滯和DNA損傷誘導基因153),以啟動促凋亡途徑[23](圖1)。目前,已有研究表明,ERS可以在轉錄及翻譯水平上調MMP-2的表達[17],并且MMP-2在鞏膜重塑的發生中占有重要地位。除此之外,2017年陳慶中[24]發現了ERS在豚鼠FDM模型中與轉化生長因子(TGF)-β1交互作用從而調節鞏膜重塑的發生發展。2022年唐曉蘭等[25]證明了ERS可以通過調節凋亡蛋白的表達以調節鞏膜重塑。與此同時,日本學者Ikeda等[6]對鏡片誘導性近視(LIM)模型小鼠腹腔內注射及局部使用4-苯基丁酸抑制ERS,并與使用了磷酸鹽緩沖液的模型小鼠進行對比,發現ERS被抑制后眼軸增長受到了有效的抑制。雖然尚無研究證明UPR三條分支在鞏膜重塑發生中的具體變化及影響機制,但是我們仍認為,UPR是未來研究鞏膜重塑發生機制的重要方向。
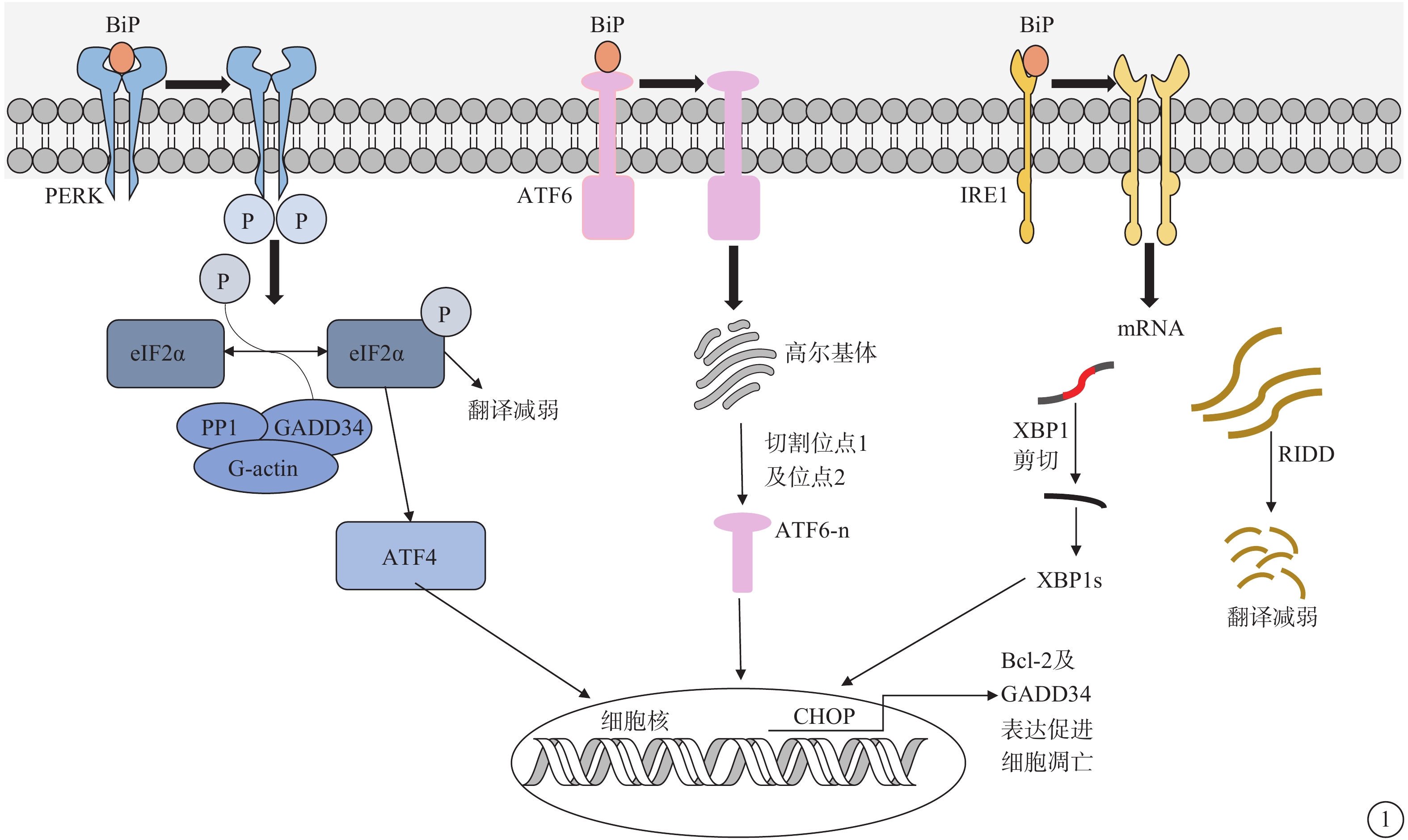
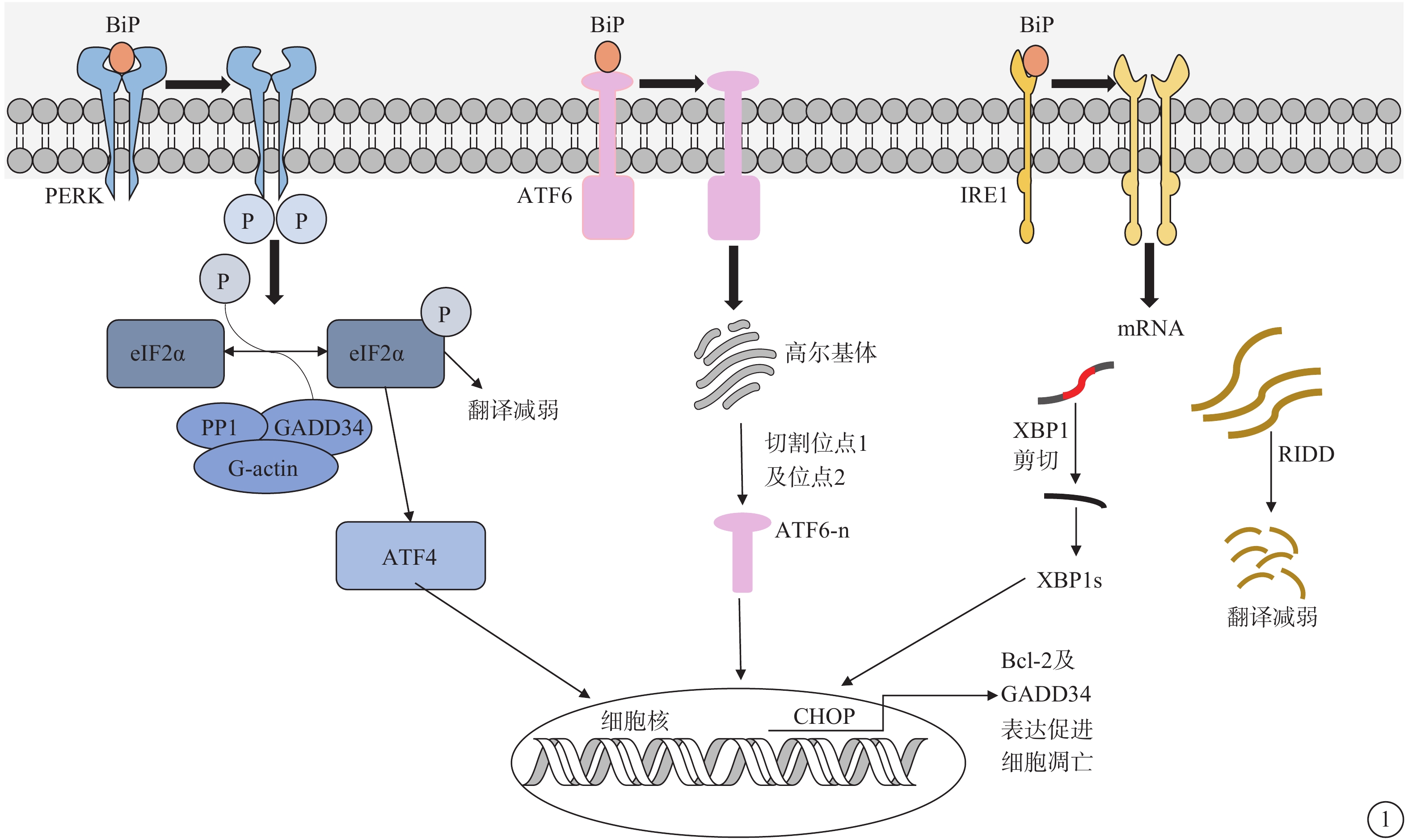 圖1
ERS作用示意圖 ERS:內質網應激;PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;IRE1:肌醇需求酶-1;ATF:轉錄激活因子;RIDD:IRE1依賴性衰減;eIF2α:真核細胞起始因子2α;GADD34:生長阻滯和DNA損傷誘導因子34;PP1:蛋白磷酸酶-1;G-actin:G-肌動蛋白;XBP1:X盒結合蛋白-1;BCL-2:抗凋亡蛋白;CHOP:CCAAT/增強子結合同源蛋白。內質網腔內的未折疊蛋白積累,BiP與PERK、IRE1以及ATF6解離,導致折疊蛋白反應(UPR)發生。活性IRE1通過轉錄因子XBP1的非常規剪接上調UPR靶基因。IRE1在ERS過程中通過降解內質網定位的mRNA轉錄物減少分泌蛋白的翻譯,這一過程被稱為RIDD。ATF6的激活導致其轉運到高爾基體,在那里被蛋白水解處理。XBP1和ATF6-n上調編碼內質網折疊和降解機制的基因。激活的PERK使eIF2α磷酸化,同時,eIF2α的磷酸化誘導轉錄因子ATF4的翻譯,促進氧化折疊和氨基酸合成。此外,ATF4還可以通過上調GADD34促進正常翻譯速率的恢復
圖1
ERS作用示意圖 ERS:內質網應激;PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;IRE1:肌醇需求酶-1;ATF:轉錄激活因子;RIDD:IRE1依賴性衰減;eIF2α:真核細胞起始因子2α;GADD34:生長阻滯和DNA損傷誘導因子34;PP1:蛋白磷酸酶-1;G-actin:G-肌動蛋白;XBP1:X盒結合蛋白-1;BCL-2:抗凋亡蛋白;CHOP:CCAAT/增強子結合同源蛋白。內質網腔內的未折疊蛋白積累,BiP與PERK、IRE1以及ATF6解離,導致折疊蛋白反應(UPR)發生。活性IRE1通過轉錄因子XBP1的非常規剪接上調UPR靶基因。IRE1在ERS過程中通過降解內質網定位的mRNA轉錄物減少分泌蛋白的翻譯,這一過程被稱為RIDD。ATF6的激活導致其轉運到高爾基體,在那里被蛋白水解處理。XBP1和ATF6-n上調編碼內質網折疊和降解機制的基因。激活的PERK使eIF2α磷酸化,同時,eIF2α的磷酸化誘導轉錄因子ATF4的翻譯,促進氧化折疊和氨基酸合成。此外,ATF4還可以通過上調GADD34促進正常翻譯速率的恢復
2.1 IRE1-X盒結合蛋白-1(XBP1)通路
IRE1是UPR最保守的信號轉導分子之一,它是一種位于內質網膜上的絲氨酸/蘇氨酸激酶,參與維持內質網穩態[26]。當ERS發生時,與BiP解離的活性IRE1催化XBP1 mRNA中26個核苷酸內含子的切除,導致密碼子閱讀框架的改變并產生一種活性穩定的轉錄因子XBP1[27-28]。XBP1是一種特殊的轉錄因子,可以激活廣泛的UPR靶基因的表達,這些基因涉及蛋白質折疊(伴侶和折疊酶)、蛋白質進入內質網、蛋白質質量控制以及其他調節糖基化和內質網相關降解的因素[29-30]。除了以上述方式減少關鍵的蛋白質折疊成分外,IRE1的核糖核酸酶還通過調節IRE1依賴性衰變(RIDD)來調節mRNA和microRNA的穩定性[31-32]。這是一種保守的機制,通過降解mRNA來減少內質網內的蛋白質負荷,從而維持內質網穩態的動態平衡。此外,RIDD還參與各種細胞過程的調節,包括細胞死亡和炎癥反應[33]。最后,持續的IRE1刺激會激活凋亡信號調節激酶1(A-SK1)致使下游的c-jun氨基末端激酶(JNK)磷酸化[34],并在ERS未停止的情況下促進細胞凋亡。據報道,磷酸化的JNK既能促進促凋亡基因(BAX)的表達,又能抑制抗凋亡基因(BCL-2)的表達 [30]。BCL-2家族不僅包含了促凋亡基因同時也含有抗凋亡基因,其通過調節線粒體膜外膜的完整性來調控這種包括ERS在內的“內在”的凋亡途徑。既往研究發現了IRE1在脂肪和膽固醇生物生成、肝臟藥物解毒、腸道炎癥、免疫細胞分化和活動、胰腺功能(內分泌和外分泌功能)和腦生理等方面的作用[35]。這說明IRE1在全身器官保持動態平衡的過程中起到了重要作用。但是,關于IRE1與鞏膜重塑之間的聯系罕有報道。2022年唐曉蘭等[25]利用體外培養的人胚胎眼鞏膜成纖維細胞,模擬近視鞏膜缺氧環境,發現缺氧可以導致IRE1表達上調,同時其磷酸化-IRE1水平也隨缺氧時間延長逐漸升高,且隨著缺氧時間的延長,細胞凋亡率逐漸增加。這證明了缺氧可以誘發ERS,長時間的ERS通過持續激活IRE1的磷酸激酶活性,致使JNK磷酸化激活。該研究同時發現了在缺氧條件下BCL-2表達下調、BAX表達明顯上調以及成纖維細胞轉分化標志物表達升高,進一步證明缺氧激活了ERS反應并參與調節缺氧狀態下鞏膜成纖維細胞的狀態(圖2)。
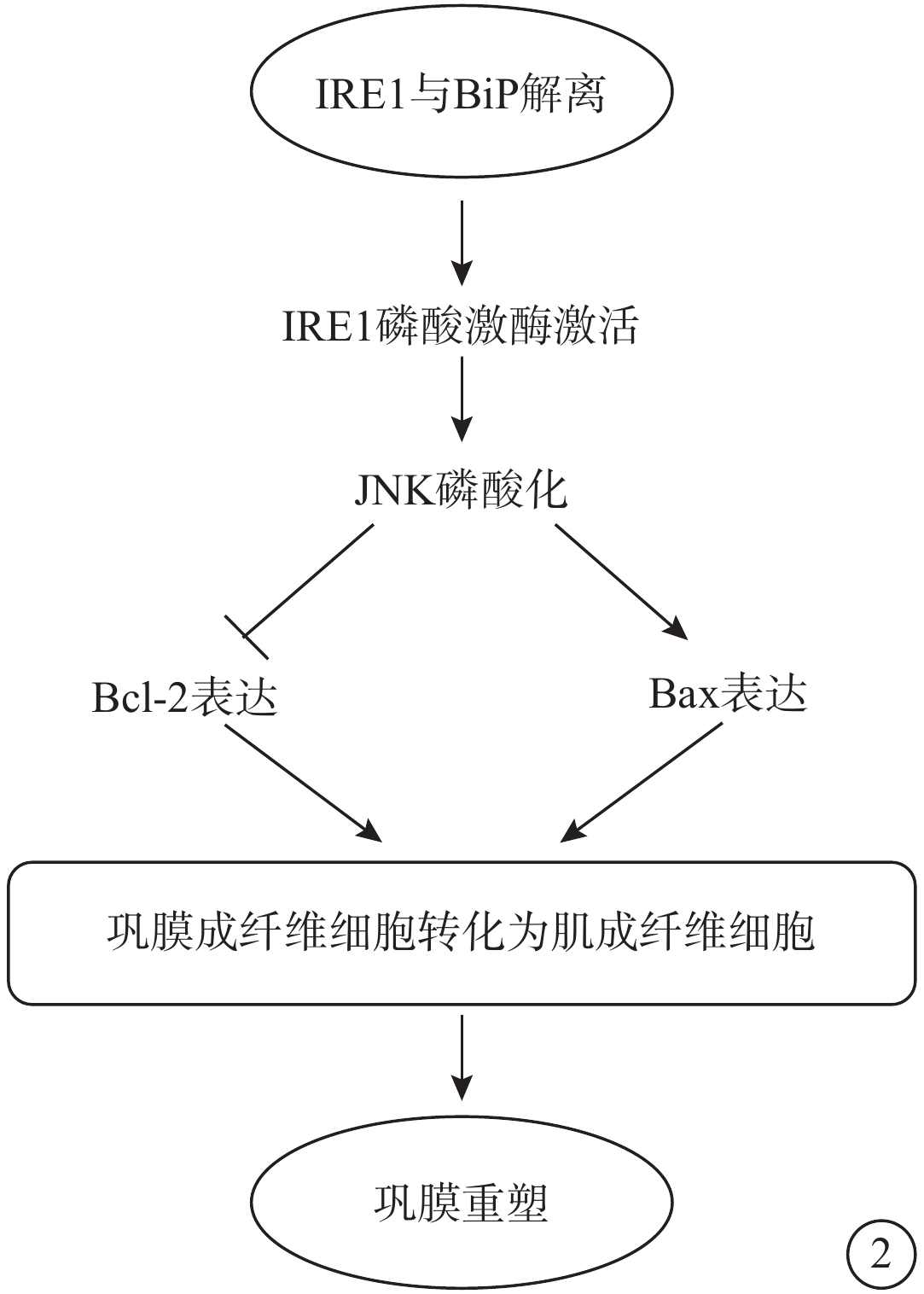
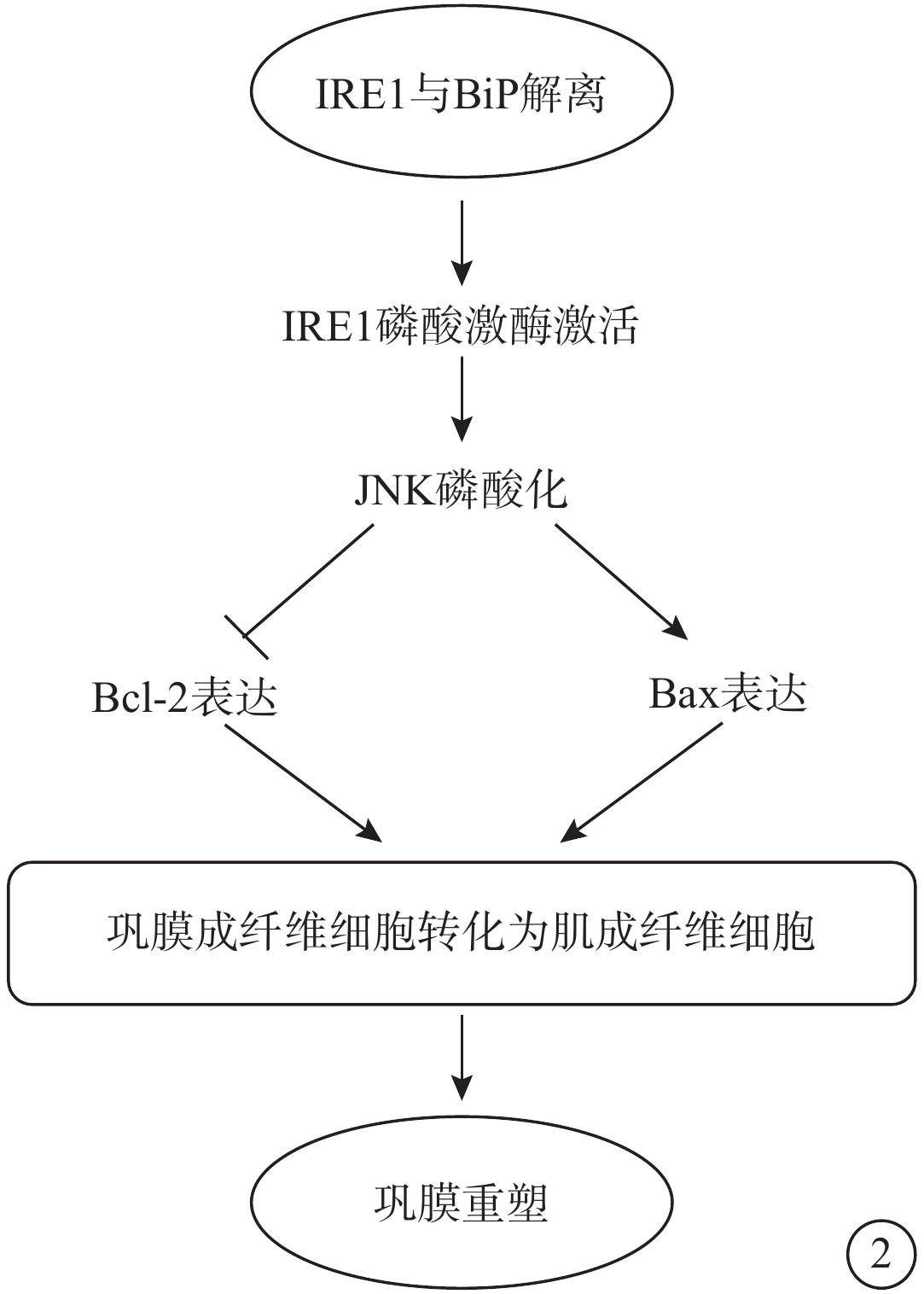 圖2
IRE1通路示意圖 IRE1:肌醇需求酶-1;BiP:免疫球蛋白重鏈結合蛋白;JNK:c-jun氨基末端激酶;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白。長時間的內質網應激通過持續激活IRE1的磷酸激酶活性,致使JNK磷酸化激活,從而促進抗凋亡基因BCL-2表達下調、促凋亡基因BAX表達明顯上調并參與促進鞏膜成纖維細胞轉化為肌成纖維細胞導致鞏膜重塑
圖2
IRE1通路示意圖 IRE1:肌醇需求酶-1;BiP:免疫球蛋白重鏈結合蛋白;JNK:c-jun氨基末端激酶;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白。長時間的內質網應激通過持續激活IRE1的磷酸激酶活性,致使JNK磷酸化激活,從而促進抗凋亡基因BCL-2表達下調、促凋亡基因BAX表達明顯上調并參與促進鞏膜成纖維細胞轉化為肌成纖維細胞導致鞏膜重塑
IRE1-XBP1通路作為UPR通路之一,其可以通過激活A-SK1并導致下游的JNK磷酸化來誘導細胞凋亡。并且,根據既往研究報道,該通路參與了缺氧條件下的鞏膜重塑。目前,國內外對于該通路在鞏膜重塑發生發展的具體分析研究較少,但可以確定的是,該通路的確參與了鞏膜重塑。既往研究在抑制了LIM模型中的IRE1通路后并未發現鞏膜重塑受到明顯影響。因此,我們認為,后續研究可以通過觀察LRE1抑制或敲除后的FDM及LIM模型中鞏膜重塑的發生發展,以判斷LRE1通路在鞏膜重塑的發生中所起到的具體作用,并借此尋找預防近視藥物的新方向及新靶點。雖然尚不確定IRE1激活的其他因子在鞏膜重塑發生過程中起到的作用,但IRE1依舊是值得關注的近視預防的新方向之一。
2.2 PERK-eIF2α-ATF4通路
PERK同樣是一種位于內質網膜的跨膜絲氨酸/蘇氨酸激酶,其腔結構域與IRE1同源,因此兩者有著相似的激活機制[36]。當ERS發生時,PERK與BiP解離并通過其反式自動磷酸化增加eIF2的α亞單位的酶活性同時引導其磷酸化[37],導致eIF2α活性降低,從而減緩蛋白質翻譯,給細胞充分的時間來嘗試折疊已存在于內質網管腔中的蛋白質[38]。這種現象被稱為整合應激反應[39]。與此同時,PERK同樣可以通過激活ATF4來終止整合應激反應從而恢復正常的翻譯[5]。內質網通過以上方式減弱ERS,從而減少了錯誤折疊的蛋白質數量,導致UPR信號減弱,使得細胞存活。盡管eIF2α磷酸化導致的蛋白質翻譯暫停對處于ERS下的細胞是有益的,但是,PERK的過度激活則會導致ATF4的激活,并上調CHOP的表達以抑制抗凋亡基因BCL-2的表達,同時增強促凋亡基因BAX的表達,最后導致細胞死亡[40]。近年來,部分研究發現了PERK-eIF2α-ATF4通路通過對MMP-2轉錄和翻譯調節來影響淺層絨毛外滋養細胞侵襲的發生發展[17]。同時,Ren等[41]發現了缺氧誘導因子-1α亦通過影響MMP-2的表達來改變ECM成分以促進鞏膜重塑,最終導致近視。最近,Ikeda等[6]通過藥物和遺傳方法抑制或敲除PERK發現,當PERK及ATF6同時受到抑制時,LIM模型中鞏膜重塑會受到抑制;除此之外,研究者對小鼠眼球局部用藥促進eIF2α磷酸化1周后,小鼠眼球出現了近視表現。這證明了LIM模型中ERK-eIF2α-ATF4通路參與了鞏膜重塑的調節 (圖3)。
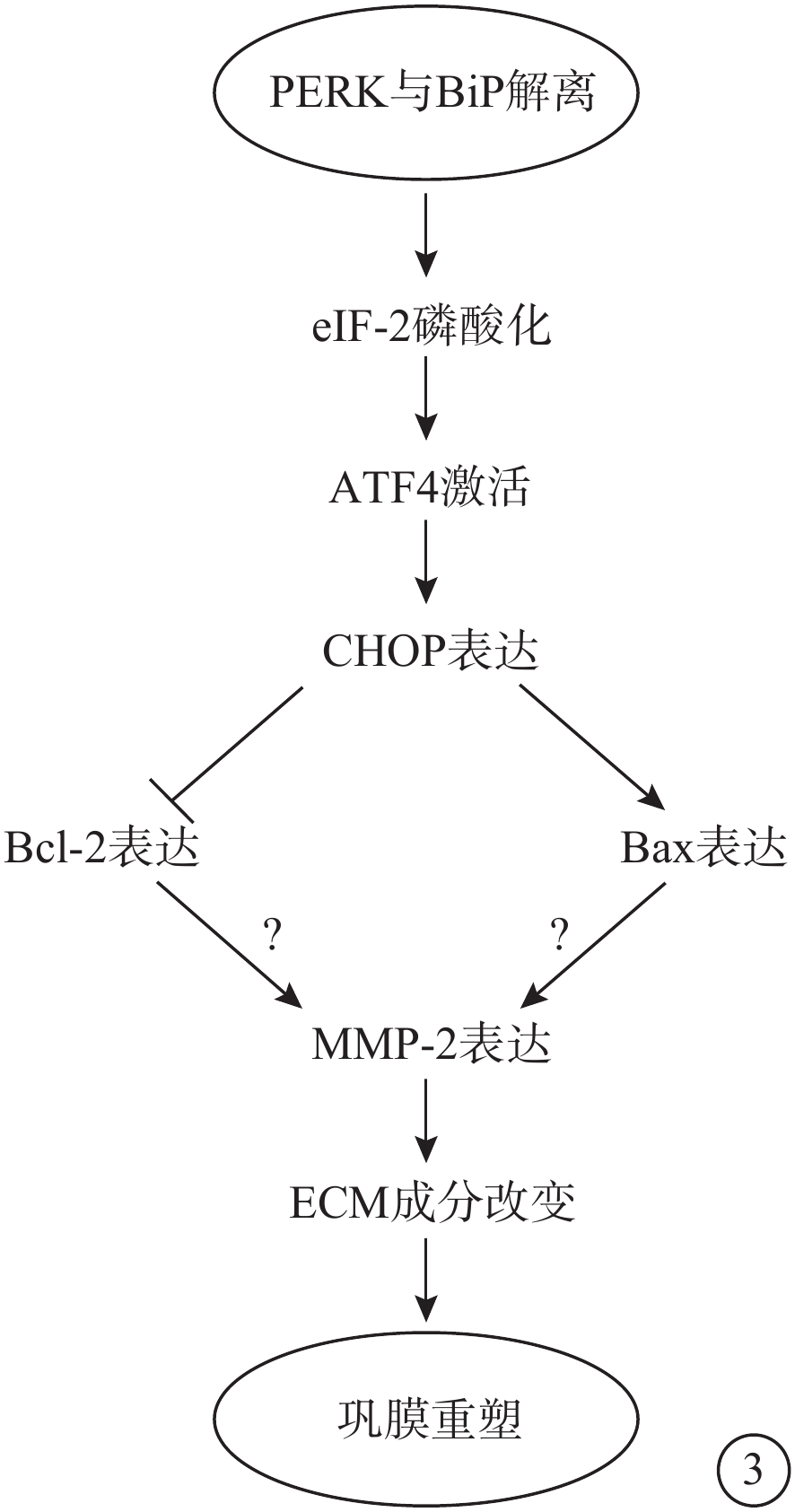
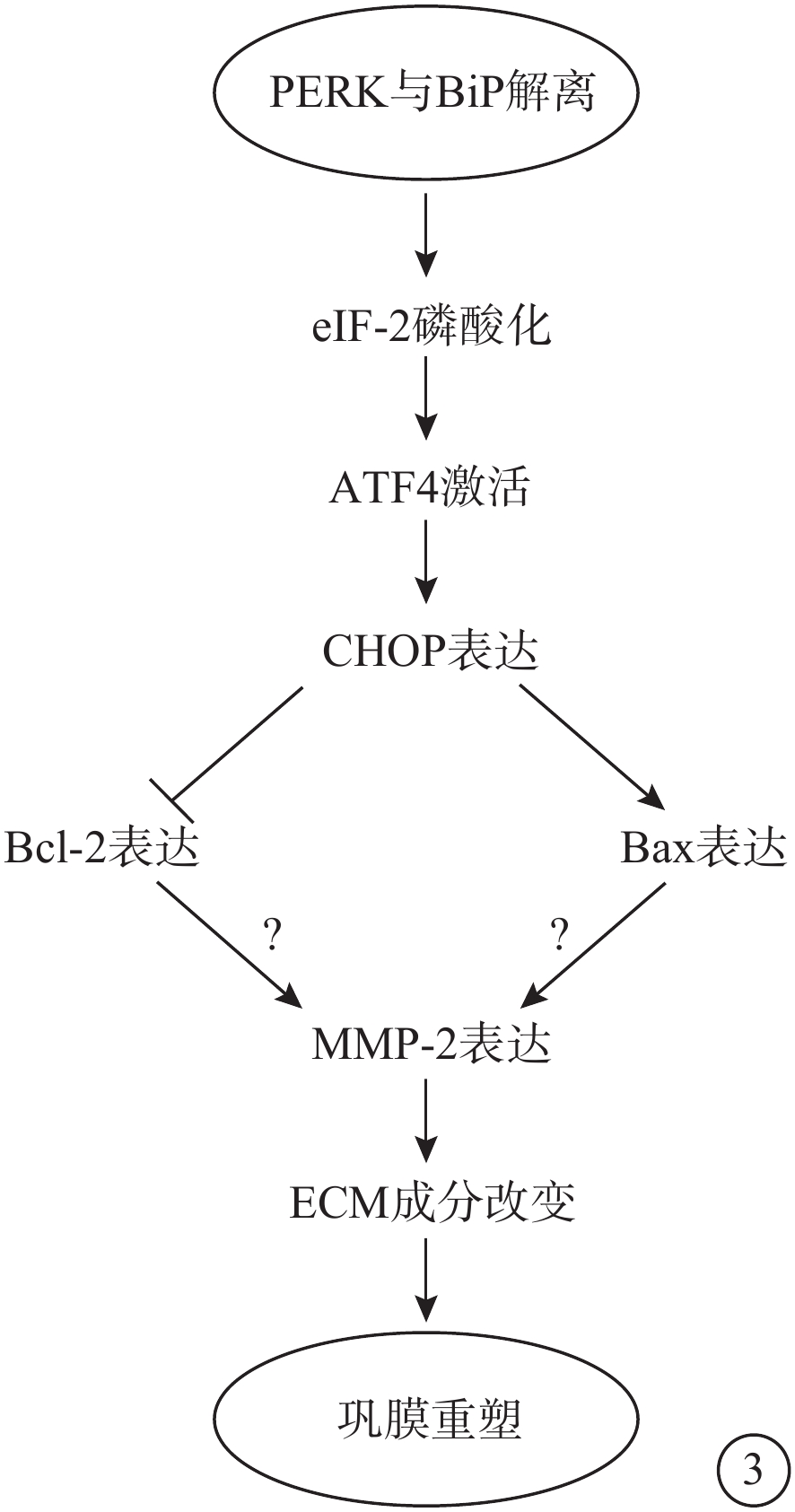 圖3
PERK通路示意圖 PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;eIF-2:翻譯起始因子-2;ATF-4:轉錄激活因子4;CHOP:CCAAT/增強子結合同源蛋白;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白;MMP-2:基質金屬蛋白酶-2;ECM:細胞外基質。PERK-eIF2α-ATF4通路通過對MMP-2的轉錄和翻譯進行調節,而MMP-2通過調節ECM成分改變促進鞏膜重塑。但目前尚無研究證明PERK-eIF2α-ATF4是否可以通過影響MMP-2導致鞏膜重塑
圖3
PERK通路示意圖 PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;eIF-2:翻譯起始因子-2;ATF-4:轉錄激活因子4;CHOP:CCAAT/增強子結合同源蛋白;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白;MMP-2:基質金屬蛋白酶-2;ECM:細胞外基質。PERK-eIF2α-ATF4通路通過對MMP-2的轉錄和翻譯進行調節,而MMP-2通過調節ECM成分改變促進鞏膜重塑。但目前尚無研究證明PERK-eIF2α-ATF4是否可以通過影響MMP-2導致鞏膜重塑
雖然尚不明確PERK通路是否通過影響MMP-2來誘發LIM相關鞏膜重塑,但根據既往研究,PERK抑制劑可有效抑制LIM導致的眼軸延長[6]。這說明該通路的研究對于未來LIM發生機制的分析及LIM的防治具有重要價值。除此之外,國內外尚無研究對FDM模型中PERK與鞏膜重塑的關系,因此,觀察PERK抑制或敲除后的FDM模型中鞏膜重塑的變化同樣是可行的研究方向。
2.3 ATF6通路
ATF6是一種內質網跨膜蛋白,目前對其的了解是最少的。它包括了一個潛在的N-末端轉錄因子、一個跨膜區和一個內質網定位的C-末端結構域[42]。當內質網內蓄積大量錯誤折疊蛋白質時,ERS導致相對分子質量為90×103的ATF6反轉錄體從內質網運輸到高爾基體。在高爾基體中,位點1和位點2的蛋白酶順序切割釋放N-末端結構域,從而產生了釋放出具有活性的相對分子質量為50×103的n端片段ATF6-n,并作為激活因子去激活細胞核內的靶基因[43],如XBP1及CHOP[44]。有趣的是,ATF6-n通過形成異二聚體與XBP1表現出串擾,這可能會驅動特定的基因表達程序,從而擴大內質網大小并增加其蛋白質折疊能力[45]。 已有研究發現,ATF6通過與侏儒相關轉錄因子(Runx2)結合促進肥大軟骨細胞分化,從而促進軟骨內骨的縱向生長(圖4),其中Runx2是成骨細胞分化和肥大軟骨細胞形成的重要中樞調節因子[46]。軟骨的縱向及軸向延長模式與鞏膜的軸向延長模式類似,但是否依賴同一種方式誘導尚不明確。Ikeda等[6]在通過藥物抑制了ATF6位點2蛋白酶的表達后,發現鞏膜重塑受到了明顯抑制;同時,其利用成簇規律間隔的短回文重復序列及其相關蛋白9系統敲除了LIM模型中的ATF6,發現眼軸的增長同樣受到了抑制;并且,同時敲除ATF6及PERK同樣可以抑制眼軸的增長。因此,可以認為ATF6與PERK聯合作用控制眼軸的增長,且ATF6可能占據主要地位。
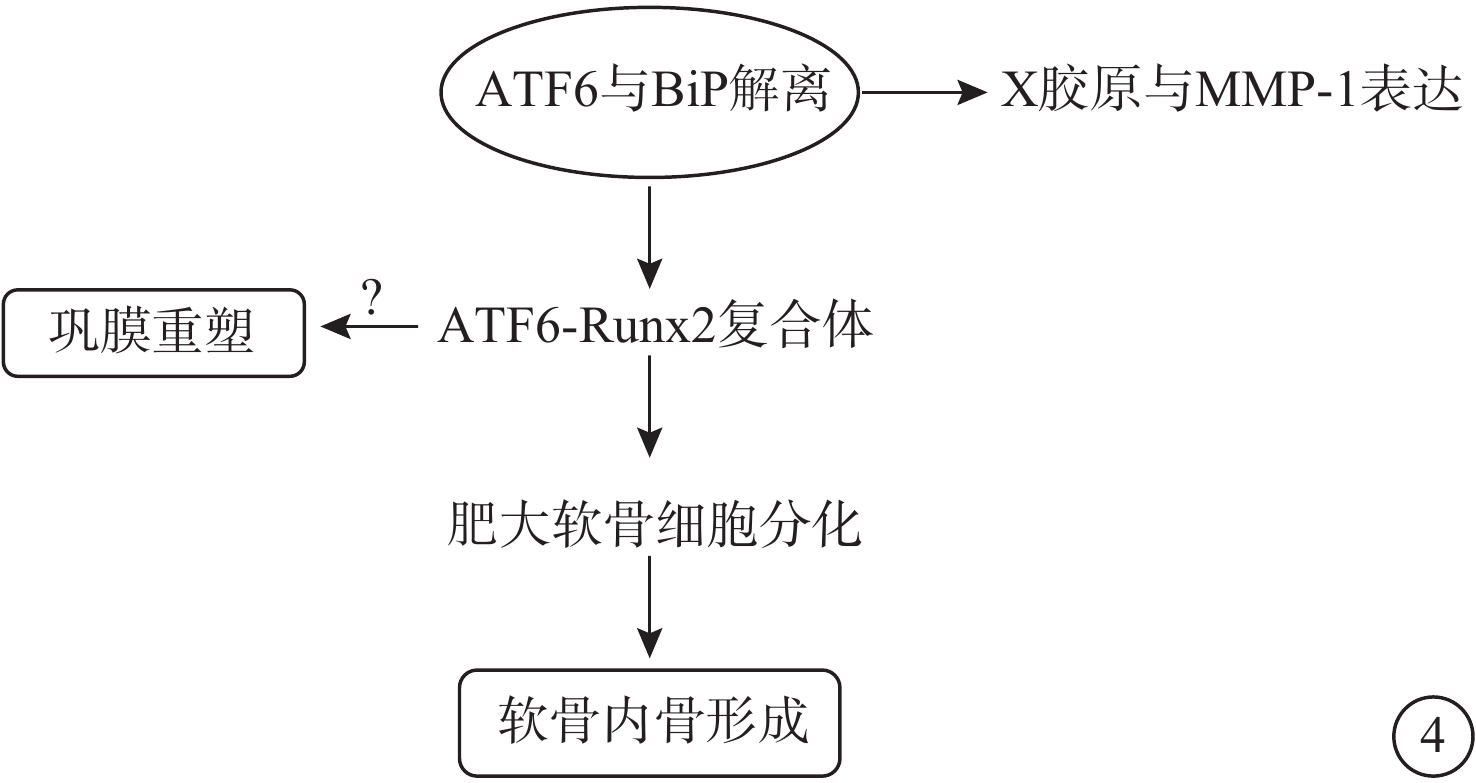
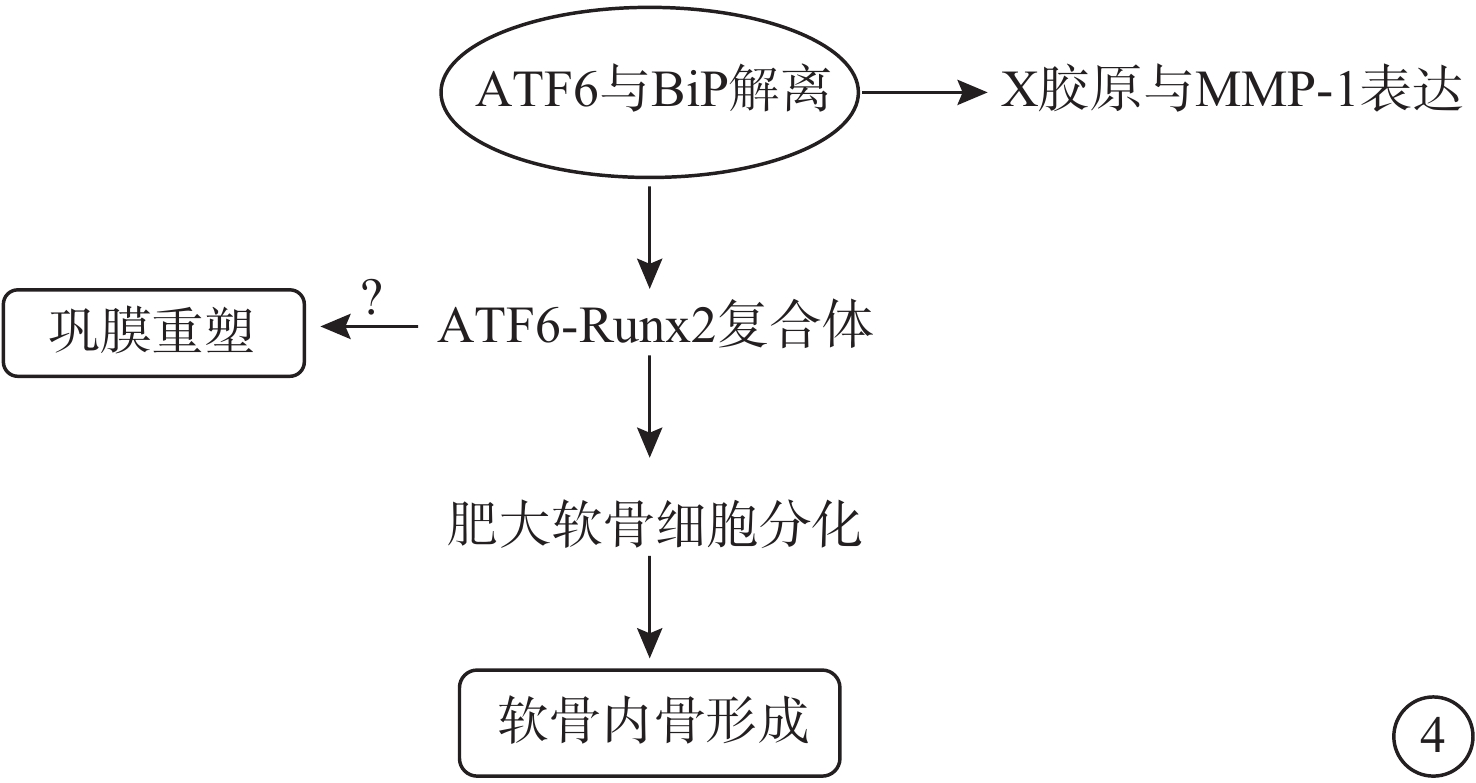 圖4
ATF6通路示意圖 ATF6:轉錄激活因子-6;BiP:免疫球蛋白重鏈結合蛋白;MMP-13:基質金屬蛋白酶-13;Runx2:侏儒相關轉錄因子。ATF6和Runx2可以在軟骨細胞分化過程中形成蛋白質復合體,然后ATF6的過表達可以促進膠原X和MMP-13的表達,進而促進依賴Runx2的肥大軟骨細胞分化。目前尚不明確ATF6是否可以通過相同方式影響鞏膜組織
圖4
ATF6通路示意圖 ATF6:轉錄激活因子-6;BiP:免疫球蛋白重鏈結合蛋白;MMP-13:基質金屬蛋白酶-13;Runx2:侏儒相關轉錄因子。ATF6和Runx2可以在軟骨細胞分化過程中形成蛋白質復合體,然后ATF6的過表達可以促進膠原X和MMP-13的表達,進而促進依賴Runx2的肥大軟骨細胞分化。目前尚不明確ATF6是否可以通過相同方式影響鞏膜組織
雖然尚不明確ATF6通路在LIM模型中調節眼軸長度的具體方式。但是,結合既往研究,我們認為與軟骨內骨成長類似,ATF6通路可能通過與Runx2結合以促進眼軸長度的改變。雖然,目前尚無研究全面分析ATF6在鞏膜重塑發生發展中的作用,但ATF6通路仍是未來研究鞏膜重塑基質的重要方向。
ERS通過激活IRE1-XBP1、PERK-eIF2α-ATF4及ATF6通路以維持蛋白質折疊的動態平衡。研究發現,MMP-2通過調節ECM的合成與降解導致鞏膜重塑的發生,是鞏膜重塑機制的熱點研究對象之一[47]。該蛋白是MMP作為前體酶原合成的,其折疊發生在內質網內[48]。因此,內質網的調控對于MMP-2的正確折疊至關重要。Lee等[17]已經報道了PERK-eIF2α-ATF4通路對MMP-2轉錄和翻譯調節作用。因此,我們仍認為,PERK-eIF2α-ATF4通路在鞏膜重塑的發生及發展中占有一席之地。因為PERK與IRE1具有相同腔結構域[36],我們猜想與PERK有著相同激活機制的IRE1可能存在通過影響MMP-2的轉錄及翻譯來影響鞏膜重塑的機制,所以研究并分析PERK-eIF2α-ATF4通路及IRE1-XBP1通路在調控MMP-2的表達中的貢獻,對于之后分析及研究鞏膜重塑干預靶點具有重要價值。除此之外,唐曉蘭等[25]發現,IRE1-XBP1通路可以通過調節CHOP的激活來控制BCL-2表達下調以及BAX表達上調,并通過此方式調控鞏膜成纖維細胞的功能。已有文獻指出,IRE1-XBP1、PERK-eIF2α-ATF4及ATF6通路均可調節CHOP的激活[24]。雖然目前尚無研究證明三條通路均可通過調節CHOP的激活來調控鞏膜成纖維細胞的功能以及三者在此過程中發揮的作用大小,但是,既往研究結果提示這應是未來研究鞏膜重塑發生發展的重要方向。與此同時,目前對于ATF6通路的研究報道雖然較少,但是考慮到其在骨組織中的重要作用,該通路亦應是是未來分析及闡述鞏膜重塑機制的重要切入點。
3 總結與展望
ERS作為調控細胞內蛋白質折疊的細胞器,其對細胞穩定性的作用不言而喻。目前,國內外眾多研究發現了ERS中的三條通路與近視相關鞏膜重塑有著千絲萬縷的聯系。我們認為這三條通路對于日后分析及研究鞏膜重塑機制及鞏膜重塑的干預手段具有價值。隨著大數據時代的到來,數字化分析及信息共享為科研工作者提供了極大的便利,通過將大數據與LIM及FDM模型相結合的方式,未來對于鞏膜重塑的研究將會有更廣闊的空間。
近年來,近視的患病率不斷上升,已成為全球的公共衛生問題[1-2]。導致近視的主要原因是眼軸長度的改變,這一過程是鞏膜重塑的結果。一般認為,近視的發生發展與鞏膜重塑密切相關,鞏膜重塑是一個動態過程,其涉及了鞏膜細胞外基質(ECM)的合成與降解[3]。為了有效干預近視的進一步發生發展,分析及闡明鞏膜重塑的具體機制至關重要。已有研究證明,內質網應激(ERS)在維持細胞穩態和組織穩定性、纖維化疾病ECM重塑的發生發展以及近視相關鞏膜重塑中發揮重要作用[4-6]。但對于ERS與ECM重塑的綜合分析國內外尚無報道。現就ERS在鞏膜重塑中的研究現狀及進展作一綜述,以期為后續鞏膜重塑機制的分析研究提供參考。
1 鞏膜重塑
鞏膜是致密的纖維粘彈性結締組織,對維持眼球的形狀起著關鍵作用。其主要結構成分是由鞏膜成纖維細胞分泌的膠原纖維,其中Ⅰ型膠原蛋白構成的膠原纖維占比最高[7]。除此之外,Ⅰ型膠原蛋白亦是鞏膜ECM的主要組成成分[3]。鞏膜的胚胎發育伴隨著鞏膜成纖維細胞的生長和繁殖、膠原等的分泌、膠原類型和含量的變化以及膠原纖維的形成等[8]。膠原蛋白形成膠原纖維,使鞏膜具有很強的彈性,并保持鞏膜結構和眼球的形狀[9]。因此,膠原蛋白是研究鞏膜重塑的關鍵蛋白。在近視發生過程中,鞏膜是靶組織,而膠原蛋白則是所有調節機制的靶蛋白。近視發生的早期,鞏膜的Ⅰ型膠原蛋白mRNA水平降低,導致Ⅰ型膠原蛋白減少、鞏膜ECM成分改變,從而引起鞏膜變薄、眼軸增長及眼球的形態改變[10-11]。因此,鞏膜重塑的實質其實是鞏膜ECM的重塑,主要包括ECM mRNA表達和鞏膜生物力學性質的變化[12]。其中,膠原纖維的直徑、分布和方向都會影響鞏膜的生物力學特性。
目前,大量研究表明,鞏膜重塑的發生與脈絡膜變薄、血流減少密切相關[13]。脈絡膜是鞏膜血液供應的主要來源,其血流量的減少可能導致鞏膜缺氧,采用單細胞測序分析技術也發現近視眼的鞏膜中缺氧信號富集,由此證明近視的發生發展過程中鞏膜確實存在著缺氧現象[14]。
內質網是調控細胞內蛋白質正確折疊的重要細胞器,只有適當折疊的蛋白質才能被運送到高爾基體進行進一步加工[15]。當出現蛋白質過載、折疊能力減弱或者病理性刺激如缺氧時則可能誘發ERS并激活未折疊蛋白反應(UPR)[16]。目前,已有研究表明,ERS可以在轉錄及翻譯水平上調基質金屬蛋白酶(MMP)-2的表達[17]。這與既往研究發現的缺氧可以導致MMP-2上調以及形覺剝奪性近視(FDM)小鼠模型中MMP-2表達明顯升高相一致[18]。MMP-2可以降解鞏膜Ⅰ型膠原蛋白,導致膠原變薄、ECM成分改變從而導致鞏膜重塑并引起近視的發生[18]。這說明ERS在鞏膜重塑的發生中占有重要地位。
2 ERS
內質網是真核細胞中最大的細胞器之一,其在細胞中三分之一以上蛋白質的合成、折疊和結構成熟過程中均起著重要作用[19]。當蛋白質負荷和折疊能力之間的不平衡、營養缺乏以及缺氧等刺激因素出現時,作為回應,細胞激活了ERS并表現為一種稱為UPR的適應性機制[20]。UPR通過抑制蛋白質翻譯、誘導內質網相關分子伴侶促進未折疊蛋白質的折疊、激活內質網相關蛋白降解系統去除未折疊蛋白質以及促進細胞存活來恢復蛋白質的動態平衡。
UPR有三個分支,由位于內質網膜上的不同的ERS傳感器啟動,其分別是蛋白激酶RNA (PKR)樣內質網激酶(PERK)、肌醇需求酶-1(IRE1)以及轉錄激活因子-6(ATF6)。在生理情況下,這些蛋白質與ER伴侶蛋白即免疫球蛋白重鏈結合蛋白(BiP,也稱為GRP78)結合,并保持無活性狀態。當ERS發生時,BiP與上述三種傳感器蛋白解離,并觸發了UPR的三條分支[21]。UPR的三條分支均會通過蛋白質折疊需求和能力,重新調整到動態平衡,以便細胞能夠繼續生存和發揮功能[22]。然而,當長期慢性的ERS難以恢復內質網的正常功能時,UPR的三條分支將會通過激活CCAAT/增強子結合同源蛋白(CHOP,也稱為生長停滯和DNA損傷誘導基因153),以啟動促凋亡途徑[23](圖1)。目前,已有研究表明,ERS可以在轉錄及翻譯水平上調MMP-2的表達[17],并且MMP-2在鞏膜重塑的發生中占有重要地位。除此之外,2017年陳慶中[24]發現了ERS在豚鼠FDM模型中與轉化生長因子(TGF)-β1交互作用從而調節鞏膜重塑的發生發展。2022年唐曉蘭等[25]證明了ERS可以通過調節凋亡蛋白的表達以調節鞏膜重塑。與此同時,日本學者Ikeda等[6]對鏡片誘導性近視(LIM)模型小鼠腹腔內注射及局部使用4-苯基丁酸抑制ERS,并與使用了磷酸鹽緩沖液的模型小鼠進行對比,發現ERS被抑制后眼軸增長受到了有效的抑制。雖然尚無研究證明UPR三條分支在鞏膜重塑發生中的具體變化及影響機制,但是我們仍認為,UPR是未來研究鞏膜重塑發生機制的重要方向。
圖1
ERS作用示意圖 ERS:內質網應激;PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;IRE1:肌醇需求酶-1;ATF:轉錄激活因子;RIDD:IRE1依賴性衰減;eIF2α:真核細胞起始因子2α;GADD34:生長阻滯和DNA損傷誘導因子34;PP1:蛋白磷酸酶-1;G-actin:G-肌動蛋白;XBP1:X盒結合蛋白-1;BCL-2:抗凋亡蛋白;CHOP:CCAAT/增強子結合同源蛋白。內質網腔內的未折疊蛋白積累,BiP與PERK、IRE1以及ATF6解離,導致折疊蛋白反應(UPR)發生。活性IRE1通過轉錄因子XBP1的非常規剪接上調UPR靶基因。IRE1在ERS過程中通過降解內質網定位的mRNA轉錄物減少分泌蛋白的翻譯,這一過程被稱為RIDD。ATF6的激活導致其轉運到高爾基體,在那里被蛋白水解處理。XBP1和ATF6-n上調編碼內質網折疊和降解機制的基因。激活的PERK使eIF2α磷酸化,同時,eIF2α的磷酸化誘導轉錄因子ATF4的翻譯,促進氧化折疊和氨基酸合成。此外,ATF4還可以通過上調GADD34促進正常翻譯速率的恢復
2.1 IRE1-X盒結合蛋白-1(XBP1)通路
IRE1是UPR最保守的信號轉導分子之一,它是一種位于內質網膜上的絲氨酸/蘇氨酸激酶,參與維持內質網穩態[26]。當ERS發生時,與BiP解離的活性IRE1催化XBP1 mRNA中26個核苷酸內含子的切除,導致密碼子閱讀框架的改變并產生一種活性穩定的轉錄因子XBP1[27-28]。XBP1是一種特殊的轉錄因子,可以激活廣泛的UPR靶基因的表達,這些基因涉及蛋白質折疊(伴侶和折疊酶)、蛋白質進入內質網、蛋白質質量控制以及其他調節糖基化和內質網相關降解的因素[29-30]。除了以上述方式減少關鍵的蛋白質折疊成分外,IRE1的核糖核酸酶還通過調節IRE1依賴性衰變(RIDD)來調節mRNA和microRNA的穩定性[31-32]。這是一種保守的機制,通過降解mRNA來減少內質網內的蛋白質負荷,從而維持內質網穩態的動態平衡。此外,RIDD還參與各種細胞過程的調節,包括細胞死亡和炎癥反應[33]。最后,持續的IRE1刺激會激活凋亡信號調節激酶1(A-SK1)致使下游的c-jun氨基末端激酶(JNK)磷酸化[34],并在ERS未停止的情況下促進細胞凋亡。據報道,磷酸化的JNK既能促進促凋亡基因(BAX)的表達,又能抑制抗凋亡基因(BCL-2)的表達 [30]。BCL-2家族不僅包含了促凋亡基因同時也含有抗凋亡基因,其通過調節線粒體膜外膜的完整性來調控這種包括ERS在內的“內在”的凋亡途徑。既往研究發現了IRE1在脂肪和膽固醇生物生成、肝臟藥物解毒、腸道炎癥、免疫細胞分化和活動、胰腺功能(內分泌和外分泌功能)和腦生理等方面的作用[35]。這說明IRE1在全身器官保持動態平衡的過程中起到了重要作用。但是,關于IRE1與鞏膜重塑之間的聯系罕有報道。2022年唐曉蘭等[25]利用體外培養的人胚胎眼鞏膜成纖維細胞,模擬近視鞏膜缺氧環境,發現缺氧可以導致IRE1表達上調,同時其磷酸化-IRE1水平也隨缺氧時間延長逐漸升高,且隨著缺氧時間的延長,細胞凋亡率逐漸增加。這證明了缺氧可以誘發ERS,長時間的ERS通過持續激活IRE1的磷酸激酶活性,致使JNK磷酸化激活。該研究同時發現了在缺氧條件下BCL-2表達下調、BAX表達明顯上調以及成纖維細胞轉分化標志物表達升高,進一步證明缺氧激活了ERS反應并參與調節缺氧狀態下鞏膜成纖維細胞的狀態(圖2)。
圖2
IRE1通路示意圖 IRE1:肌醇需求酶-1;BiP:免疫球蛋白重鏈結合蛋白;JNK:c-jun氨基末端激酶;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白。長時間的內質網應激通過持續激活IRE1的磷酸激酶活性,致使JNK磷酸化激活,從而促進抗凋亡基因BCL-2表達下調、促凋亡基因BAX表達明顯上調并參與促進鞏膜成纖維細胞轉化為肌成纖維細胞導致鞏膜重塑
IRE1-XBP1通路作為UPR通路之一,其可以通過激活A-SK1并導致下游的JNK磷酸化來誘導細胞凋亡。并且,根據既往研究報道,該通路參與了缺氧條件下的鞏膜重塑。目前,國內外對于該通路在鞏膜重塑發生發展的具體分析研究較少,但可以確定的是,該通路的確參與了鞏膜重塑。既往研究在抑制了LIM模型中的IRE1通路后并未發現鞏膜重塑受到明顯影響。因此,我們認為,后續研究可以通過觀察LRE1抑制或敲除后的FDM及LIM模型中鞏膜重塑的發生發展,以判斷LRE1通路在鞏膜重塑的發生中所起到的具體作用,并借此尋找預防近視藥物的新方向及新靶點。雖然尚不確定IRE1激活的其他因子在鞏膜重塑發生過程中起到的作用,但IRE1依舊是值得關注的近視預防的新方向之一。
2.2 PERK-eIF2α-ATF4通路
PERK同樣是一種位于內質網膜的跨膜絲氨酸/蘇氨酸激酶,其腔結構域與IRE1同源,因此兩者有著相似的激活機制[36]。當ERS發生時,PERK與BiP解離并通過其反式自動磷酸化增加eIF2的α亞單位的酶活性同時引導其磷酸化[37],導致eIF2α活性降低,從而減緩蛋白質翻譯,給細胞充分的時間來嘗試折疊已存在于內質網管腔中的蛋白質[38]。這種現象被稱為整合應激反應[39]。與此同時,PERK同樣可以通過激活ATF4來終止整合應激反應從而恢復正常的翻譯[5]。內質網通過以上方式減弱ERS,從而減少了錯誤折疊的蛋白質數量,導致UPR信號減弱,使得細胞存活。盡管eIF2α磷酸化導致的蛋白質翻譯暫停對處于ERS下的細胞是有益的,但是,PERK的過度激活則會導致ATF4的激活,并上調CHOP的表達以抑制抗凋亡基因BCL-2的表達,同時增強促凋亡基因BAX的表達,最后導致細胞死亡[40]。近年來,部分研究發現了PERK-eIF2α-ATF4通路通過對MMP-2轉錄和翻譯調節來影響淺層絨毛外滋養細胞侵襲的發生發展[17]。同時,Ren等[41]發現了缺氧誘導因子-1α亦通過影響MMP-2的表達來改變ECM成分以促進鞏膜重塑,最終導致近視。最近,Ikeda等[6]通過藥物和遺傳方法抑制或敲除PERK發現,當PERK及ATF6同時受到抑制時,LIM模型中鞏膜重塑會受到抑制;除此之外,研究者對小鼠眼球局部用藥促進eIF2α磷酸化1周后,小鼠眼球出現了近視表現。這證明了LIM模型中ERK-eIF2α-ATF4通路參與了鞏膜重塑的調節 (圖3)。
圖3
PERK通路示意圖 PERK:蛋白激酶RNA樣內質網激酶;BiP:免疫球蛋白重鏈結合蛋白;eIF-2:翻譯起始因子-2;ATF-4:轉錄激活因子4;CHOP:CCAAT/增強子結合同源蛋白;BCL-2:抗凋亡蛋白;BAX:促凋亡蛋白;MMP-2:基質金屬蛋白酶-2;ECM:細胞外基質。PERK-eIF2α-ATF4通路通過對MMP-2的轉錄和翻譯進行調節,而MMP-2通過調節ECM成分改變促進鞏膜重塑。但目前尚無研究證明PERK-eIF2α-ATF4是否可以通過影響MMP-2導致鞏膜重塑
雖然尚不明確PERK通路是否通過影響MMP-2來誘發LIM相關鞏膜重塑,但根據既往研究,PERK抑制劑可有效抑制LIM導致的眼軸延長[6]。這說明該通路的研究對于未來LIM發生機制的分析及LIM的防治具有重要價值。除此之外,國內外尚無研究對FDM模型中PERK與鞏膜重塑的關系,因此,觀察PERK抑制或敲除后的FDM模型中鞏膜重塑的變化同樣是可行的研究方向。
2.3 ATF6通路
ATF6是一種內質網跨膜蛋白,目前對其的了解是最少的。它包括了一個潛在的N-末端轉錄因子、一個跨膜區和一個內質網定位的C-末端結構域[42]。當內質網內蓄積大量錯誤折疊蛋白質時,ERS導致相對分子質量為90×103的ATF6反轉錄體從內質網運輸到高爾基體。在高爾基體中,位點1和位點2的蛋白酶順序切割釋放N-末端結構域,從而產生了釋放出具有活性的相對分子質量為50×103的n端片段ATF6-n,并作為激活因子去激活細胞核內的靶基因[43],如XBP1及CHOP[44]。有趣的是,ATF6-n通過形成異二聚體與XBP1表現出串擾,這可能會驅動特定的基因表達程序,從而擴大內質網大小并增加其蛋白質折疊能力[45]。 已有研究發現,ATF6通過與侏儒相關轉錄因子(Runx2)結合促進肥大軟骨細胞分化,從而促進軟骨內骨的縱向生長(圖4),其中Runx2是成骨細胞分化和肥大軟骨細胞形成的重要中樞調節因子[46]。軟骨的縱向及軸向延長模式與鞏膜的軸向延長模式類似,但是否依賴同一種方式誘導尚不明確。Ikeda等[6]在通過藥物抑制了ATF6位點2蛋白酶的表達后,發現鞏膜重塑受到了明顯抑制;同時,其利用成簇規律間隔的短回文重復序列及其相關蛋白9系統敲除了LIM模型中的ATF6,發現眼軸的增長同樣受到了抑制;并且,同時敲除ATF6及PERK同樣可以抑制眼軸的增長。因此,可以認為ATF6與PERK聯合作用控制眼軸的增長,且ATF6可能占據主要地位。
圖4
ATF6通路示意圖 ATF6:轉錄激活因子-6;BiP:免疫球蛋白重鏈結合蛋白;MMP-13:基質金屬蛋白酶-13;Runx2:侏儒相關轉錄因子。ATF6和Runx2可以在軟骨細胞分化過程中形成蛋白質復合體,然后ATF6的過表達可以促進膠原X和MMP-13的表達,進而促進依賴Runx2的肥大軟骨細胞分化。目前尚不明確ATF6是否可以通過相同方式影響鞏膜組織
雖然尚不明確ATF6通路在LIM模型中調節眼軸長度的具體方式。但是,結合既往研究,我們認為與軟骨內骨成長類似,ATF6通路可能通過與Runx2結合以促進眼軸長度的改變。雖然,目前尚無研究全面分析ATF6在鞏膜重塑發生發展中的作用,但ATF6通路仍是未來研究鞏膜重塑基質的重要方向。
ERS通過激活IRE1-XBP1、PERK-eIF2α-ATF4及ATF6通路以維持蛋白質折疊的動態平衡。研究發現,MMP-2通過調節ECM的合成與降解導致鞏膜重塑的發生,是鞏膜重塑機制的熱點研究對象之一[47]。該蛋白是MMP作為前體酶原合成的,其折疊發生在內質網內[48]。因此,內質網的調控對于MMP-2的正確折疊至關重要。Lee等[17]已經報道了PERK-eIF2α-ATF4通路對MMP-2轉錄和翻譯調節作用。因此,我們仍認為,PERK-eIF2α-ATF4通路在鞏膜重塑的發生及發展中占有一席之地。因為PERK與IRE1具有相同腔結構域[36],我們猜想與PERK有著相同激活機制的IRE1可能存在通過影響MMP-2的轉錄及翻譯來影響鞏膜重塑的機制,所以研究并分析PERK-eIF2α-ATF4通路及IRE1-XBP1通路在調控MMP-2的表達中的貢獻,對于之后分析及研究鞏膜重塑干預靶點具有重要價值。除此之外,唐曉蘭等[25]發現,IRE1-XBP1通路可以通過調節CHOP的激活來控制BCL-2表達下調以及BAX表達上調,并通過此方式調控鞏膜成纖維細胞的功能。已有文獻指出,IRE1-XBP1、PERK-eIF2α-ATF4及ATF6通路均可調節CHOP的激活[24]。雖然目前尚無研究證明三條通路均可通過調節CHOP的激活來調控鞏膜成纖維細胞的功能以及三者在此過程中發揮的作用大小,但是,既往研究結果提示這應是未來研究鞏膜重塑發生發展的重要方向。與此同時,目前對于ATF6通路的研究報道雖然較少,但是考慮到其在骨組織中的重要作用,該通路亦應是是未來分析及闡述鞏膜重塑機制的重要切入點。
3 總結與展望
ERS作為調控細胞內蛋白質折疊的細胞器,其對細胞穩定性的作用不言而喻。目前,國內外眾多研究發現了ERS中的三條通路與近視相關鞏膜重塑有著千絲萬縷的聯系。我們認為這三條通路對于日后分析及研究鞏膜重塑機制及鞏膜重塑的干預手段具有價值。隨著大數據時代的到來,數字化分析及信息共享為科研工作者提供了極大的便利,通過將大數據與LIM及FDM模型相結合的方式,未來對于鞏膜重塑的研究將會有更廣闊的空間。