細胞外過量的谷氨酸可誘導N-甲基-D-天冬氨酸(NMDA)受體過度激活,造成視網膜神經節細胞(RGC)死亡和視野進行性縮窄,這些病理改變在青光眼等致盲性眼病中已得到廣泛研究并冠名為“視網膜興奮性毒性”[1]。但NMDA誘導的視網膜興奮性毒性的分子機制,特別是RNA甲基化修飾的表觀遺傳學調控機制,仍屬未知。RNA甲基化修飾是指在RNA分子不同位置上的甲基化修飾現象,包括6-甲基腺嘌呤(m6A)、5-甲基胞嘧啶(m5C)等形式[2-3]。文獻報道,RNA甲基化修飾在眼部疾病中主要表現為m6A甲基化的分布與調控異常[4-6];但在人轉錄組中,20%~30%的mRNA帶有m5C的甲基化修飾[7],提示m5C甲基化修飾在生理和病理條件下的調控作用不容忽視。目前m5C甲基化修飾在視網膜興奮性毒性中的致病機制國內外文獻均鮮見報道。因此,本研究在NMDA誘導的大鼠視網膜興奮性毒性模型中,利用高通量m5C甲基化RNA測序(MeRIP-RNA seq)檢測視網膜中轉錄本m5C甲基化的修飾水平,并輔以生物信息學分析,并初步驗證m5C甲基化修飾對基因表達的影響。現將結果報道如下。
1 材料和方法
1.1 材料
實驗動物。Sprague Dawley(SD)雄性大鼠65只,7~8周齡,體重200~250 g;分兩批購自北京維通利華公司,飼養于天津醫科大學眼科醫院無特定病原體級標準(12 h明/12 h暗循環)動物房。本研究經天津醫科大學動物倫理委員會審核通過(動物倫理批號:SYXK 2009-0001)。所有實驗操作均符合國家衛生研究院實驗室動物護理和使用指南;實驗動物使用遵循視覺與眼科研究協會有關眼科與視覺科學研究中動物使用的相關規定。
主要試劑及儀器。NMDA(美國Sigma公司);大鼠單克隆β3-微管蛋白抗體、兔抗大鼠二抗(美國Abcam公司);抗淬滅封片劑(美國Vectashield公司);RNA提取試劑盒(美國EZBioscience公司);氧化鋯研磨珠高速組織研磨儀(武漢塞維爾生物科技有限公司);共聚焦顯微鏡(德國Zeiss公司)。
1.2 確立建模的NMDA最佳玻璃體腔注射濃度
篩選建立視網膜興奮性毒性模型時NMDA的最佳玻璃體腔注射濃度。首批25只SD大鼠適應環境7 d后,隨機分為正常對照組以及NMDA 16.7、33.3、50.0、66.7 mmol/L組(每組5只)。NMDA溶解于質量分數為0.9%的生理鹽水中,濾膜(孔徑0.2 μm)過濾,制備16.7、33.3、50.0、66.7 mmol/L無菌NMDA溶液。大鼠按體重腹腔注射40 mg/kg戊巴比妥鈉全身麻醉,鹽酸奧布卡因滴眼液眼表麻醉。不同濃度NMDA組大鼠右眼(模型眼)玻璃體腔注射相應濃度3 μl NMDA,正常對照組大鼠右眼玻璃體腔注射等體積生理鹽水。已剔除虹膜出血、晶狀體受損、穿刺及拔針過程中漏液過多以及注射后出現眼部感染的大鼠。
蘇木素-伊紅(HE)染色觀察正常對照組、不同濃度NMDA組大鼠的視網膜整體結構。建模后7 d,大鼠腹腔注射過量戊巴比妥鈉深度麻醉處死。摘除眼球,置于酸性固定液中固定。常規石蠟包埋,于視神經附近沿眼球縱向進行切片,每個眼球在相似部位連續切片5張,3 μm/張。組織切片脫蠟、脫水,HE染色,光學顯微鏡下觀察拍照。采用CellSense軟件測量視網膜各層厚度,Photoshop CS6計數視網膜神經節細胞層(GCL)中細胞數量。所有操作均采用雙盲原則。依據文獻[8-9],原發性開角型青光眼患者臨床常規檢測中發現視野缺損時,已有50%的節細胞丟失;而本研究實驗結果顯示,50 mmol/L NMDA經玻璃體腔注射7 d后,大鼠GCL細胞數量減少到正常對照組的50.09%。為使動物模型再現臨床病理改變,選取玻璃體腔注射50 mmol/L NMDA,建立視網膜興奮性毒性大鼠模型。
1.3 實驗方法
第二批SD大鼠40只,隨機分為正常對照組、50 mmol/L NMDA組,每組各20只。
視覺誘發電位(VEP)檢測大鼠視神經傳導功能。建模后7 d,過夜暗適應后,正常對照組、50 mmol/L NMDA組大鼠,按體重腹腔注射40 mg/kg戊巴比妥鈉全身麻醉,右眼(模型眼)復方托吡卡胺散瞳。弱紅光下,大鼠以俯臥位固定于實驗臺上,將針狀鉑電極置于大鼠角膜,參考電極置于枕骨粗隆,地電極插入尾部皮下,按視覺電生理記錄儀說明書,給予大鼠30.0 cd·s/m2白色閃光刺激。右眼檢查時,左眼不透光眼罩完全遮蓋。記錄大鼠全視野暗適應VEP參數,包括P1波振幅和P1波潛伏期。P1波潛伏期為起點至P1波波峰時間,P1波振幅為起點至P1波峰距離。連續測量3次,取平均值。
視網膜鋪片免疫熒光染色檢測β3微管蛋白免疫熒光染色陽性細胞個數。選取正常對照組、50 mmol/L NMDA組大鼠各9只過量麻醉處死,摘除眼球,4%多聚甲醛(PFA)固定40 min,手術顯微鏡下剝離視網膜,置于含4% PFA的24孔板中,4 ℃固定過夜。磷酸鹽緩沖液(PBS)洗2次,每孔加入1 ml通透液,4 ℃孵育過夜。PBlec緩沖液洗3次。加入β3微管蛋白抗體(鼠源單抗體,1∶1 000,PBlec緩沖液稀釋),室溫孵育過夜。PBS洗3次,加入異硫氰酸熒光素(FITC)標記的二抗(兔抗鼠,1∶500,PBlec緩沖液稀釋),PBS洗3次。將僅加FITC標記的熒光二抗染色設為陰性對照。將視網膜呈放射狀切成3~4瓣,GCL向上平鋪于載玻片上,滴加少量含4',6-二脒基-2-苯基吲哚的抗熒光衰退封片劑封片。共聚焦顯微鏡下觀察拍照。將熒光信號強度高于非特異性背景,且呈現細胞形態的胞漿綠色熒光染色視為β3微管蛋白染色陽性,應用Photoshop CS6軟件計數β3微管蛋白染色陽性的細胞數。
MeRIP-RNA seq及生物信息學分析。建模后7 d,正常對照組和50.0 mmol/L NMDA組各收集3只眼,冰上迅速剝離視網膜,置于EP管中,液氮速凍,應用MeRIP-RNA測序進行m5C表觀轉錄組學分析,由上海云序生物科技有限公司完成。采用GeneJET RNA提取試劑盒提取各組標本總RNA,GenSeq m5C RNA IP試劑盒對m5C修飾的RNA進行免疫沉淀,富集m5C修飾的轉錄本。應用NEBNext Ultra II Directional RNA Library Prep試劑盒對免疫沉淀后的m5C RNA樣本進行RNA測序文庫的構建與質檢。使用Illumina HiSeq 4000測序儀測序、堿基識別和質控,產生原始測序數據,經Q30質控、去接頭,獲得高質量數據。采用Hisat2軟件(v2.0.4)將全部樣本的高質量數據匹配到參考基因組上。應用MACS軟件識別每個樣本中的甲基化基因,diffReps軟件進行差異甲基化基因的識別,以正常對照組和50.0 mmol/L NMDA組之間的差異倍數≥2和P≤0.000 1為標準,滿足此二項條件者,鑒定為差異甲基化基因;再應用公司自有程序篩選位于mRNA外顯子上的m5C甲基化峰,進行相應注釋。最后,對差異甲基化的編碼基因進行基因注釋(GO)和京都基因與基因組百科全書(KEGG)分析。
根據m5C MeRIP-RNA測序結果,選取NMDA處理后,m5C修飾水平增高(SLFN3
總RNA的提取和逆轉錄。建模后7 d,選取正常對照組、50 mmol/L NMDA組大鼠各8只,過量麻醉處死。收集視網膜,液氮速凍,-80 ℃冰箱儲存。每個視網膜置于1個1.5 ml EP管中,分別加入直徑4、3 mm的氧化鋯研磨珠1、2,500 μl裂解液作用,于高速組織研磨器中充分研磨。研磨后液體移至另一1.5 ml EP管,按Universal RNA Purification Kit試劑盒說明書,提取視網膜組織中總RNA。應用Nanodrop 2000微量分光光度計測定所得總RNA的濃度和純度。取1 μg總RNA,按照RevertAid First Strand cDNA Synthesis Kit說明書,用隨機六聚體引物,行逆轉錄,制備cDNA。
實時定量聚合酶鏈反應(qRT-PCR)檢測正常對照組、50 mmol/L NMDA組視網膜中SLFN3、PLXNB3、CD36、HIC2 mRNA相對表達量。PubMed中查詢大鼠4個目標基因(SLFN3、PLXNB3、CD36、HIC2)以及內參基因GAPDH的序列,應用Primer Express 3.0軟件設計引物序列(表1),由深圳華大基因股份有限公司合成。以4 μl cDNA為模板,加入基因的正向、反向引物各1.5 μl,以及12.5 μl EvaGreen 2×qPCR Master Mix,再加入無核酸酶的水,補足25 μl反應體系,在96孔板中進行qRT-PCR擴增。反應條件為:50 ℃孵育2 min,95 ℃變性10 min;95 ℃變性15 s,60 ℃退火和延伸1 min,40個循環;95 ℃變性15 s,60 ℃退火15 s,95 ℃反應15 s。采用2?△△Ct法計算目的基因mRNA相對表達情況。
1.4 統計學方法
采用GraphPad Prism 8軟件進行統計學分析。各組數據資料經Shapiro-Wilk檢驗呈正態分布,經Levene檢驗證實方差齊,并以均數±標準差( ±s)表示。正常對照組、不同濃度NMDA組大鼠視網膜各層厚度、GCL細胞數量比較采用單因素方差分析,組間多重比較采用Tukey post hoc檢驗;正常對照組與50 mmol/L NMDA組間β3-微管蛋白染色陽性細胞數、VEP P1波振幅和潛伏期、mRNA相對表達量比較采用雙側非配對t檢驗。P<0.05為差異有統計學意義。
±s)表示。正常對照組、不同濃度NMDA組大鼠視網膜各層厚度、GCL細胞數量比較采用單因素方差分析,組間多重比較采用Tukey post hoc檢驗;正常對照組與50 mmol/L NMDA組間β3-微管蛋白染色陽性細胞數、VEP P1波振幅和潛伏期、mRNA相對表達量比較采用雙側非配對t檢驗。P<0.05為差異有統計學意義。
2 結果
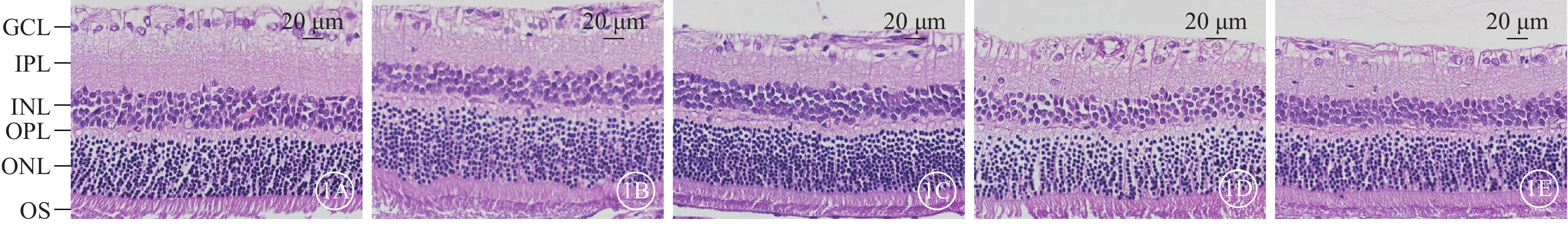
光學顯微鏡觀察發現,建模后7 d,正常對照組大鼠視網膜各層清晰,組織形態完整,細胞排列規則(圖1A)。16.7 mmol/L NMDA組大鼠視網膜GCL細胞空泡變性(圖1B);33.3 mmol/L NMDA組大鼠視網膜GCL細胞數量相對減少,排列紊亂,細胞內空泡變性(圖1C);50 mmol/L NMDA組大鼠視網膜GCL細胞數量明顯減少且伴視網膜水腫(圖1D);66.7 mmol/L NMDA組大鼠視網膜GCL細胞數量變少,核固縮,內核層變薄且紊亂,視網膜總厚度變薄(圖1E)。
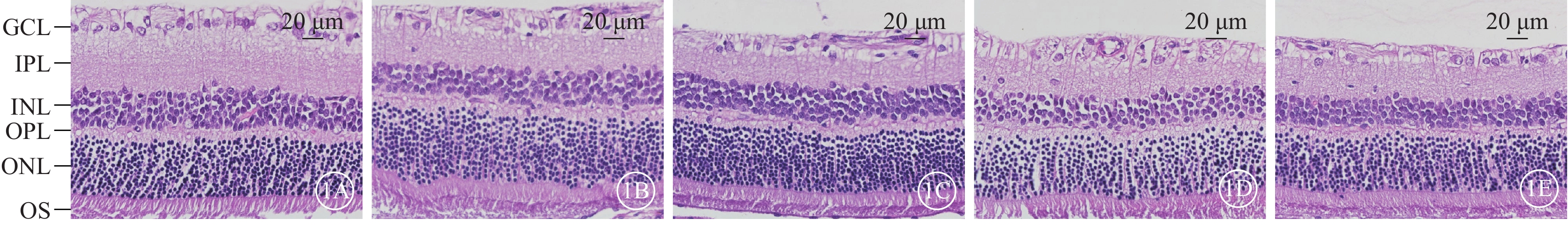 圖1
正常對照組、不同濃度NMDA組大鼠視網膜組織光學顯微鏡像 NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;IPL:內叢狀層;INL:內核層;OPL:外叢狀層;ONL:外核層;OS:光感受器細胞外節。1A示正常對照組,視網膜各層清晰,組織形態完整,細胞排列規則;1B示16.7 mmol/L NMDA組,GCL細胞空泡變性;1C示33.3 mmol/L NMDA組,GCL細胞數量相對減少,排列紊亂,細胞內空泡變性;1D示50.0 mmol/L NMDA組,GCL細胞數量明顯減少且伴視網膜水腫;1E示66.7 mmol/L NMDA組,GCL細胞數量變少,核固縮,內核層變薄且紊亂 蘇木精-伊紅染色 標尺:20 μm
圖1
正常對照組、不同濃度NMDA組大鼠視網膜組織光學顯微鏡像 NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;IPL:內叢狀層;INL:內核層;OPL:外叢狀層;ONL:外核層;OS:光感受器細胞外節。1A示正常對照組,視網膜各層清晰,組織形態完整,細胞排列規則;1B示16.7 mmol/L NMDA組,GCL細胞空泡變性;1C示33.3 mmol/L NMDA組,GCL細胞數量相對減少,排列紊亂,細胞內空泡變性;1D示50.0 mmol/L NMDA組,GCL細胞數量明顯減少且伴視網膜水腫;1E示66.7 mmol/L NMDA組,GCL細胞數量變少,核固縮,內核層變薄且紊亂 蘇木精-伊紅染色 標尺:20 μm
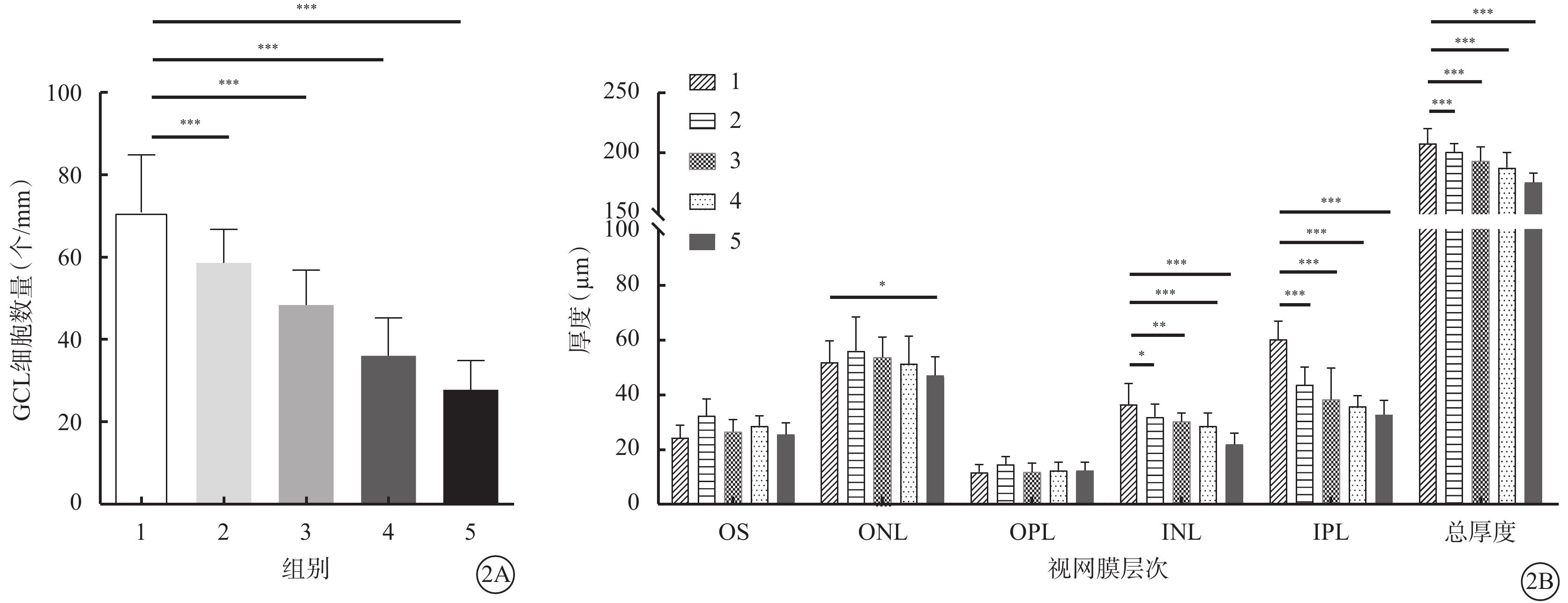
正常對照組、不同濃度NMDA組大鼠視網膜GCL細胞數量及內核層、內叢狀層、外核層、視網膜總厚度比較,差異均有統計學意義(P<0.05);外叢狀層、光感受器細胞外節厚度比較,差異無統計學意義(P>0.05)(圖2)。與正常對照組比較,16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組GCL細胞數量明顯減少,其中16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組GCL細胞數量分別為正常對照組的81.57%、67.18%、50.09%、38.64%,差異均有統計學意義(P<0.001);不同濃度NMDA組內核層厚度、視網膜總厚度均明顯降低,差異均有統計學意義(P<0.001)。
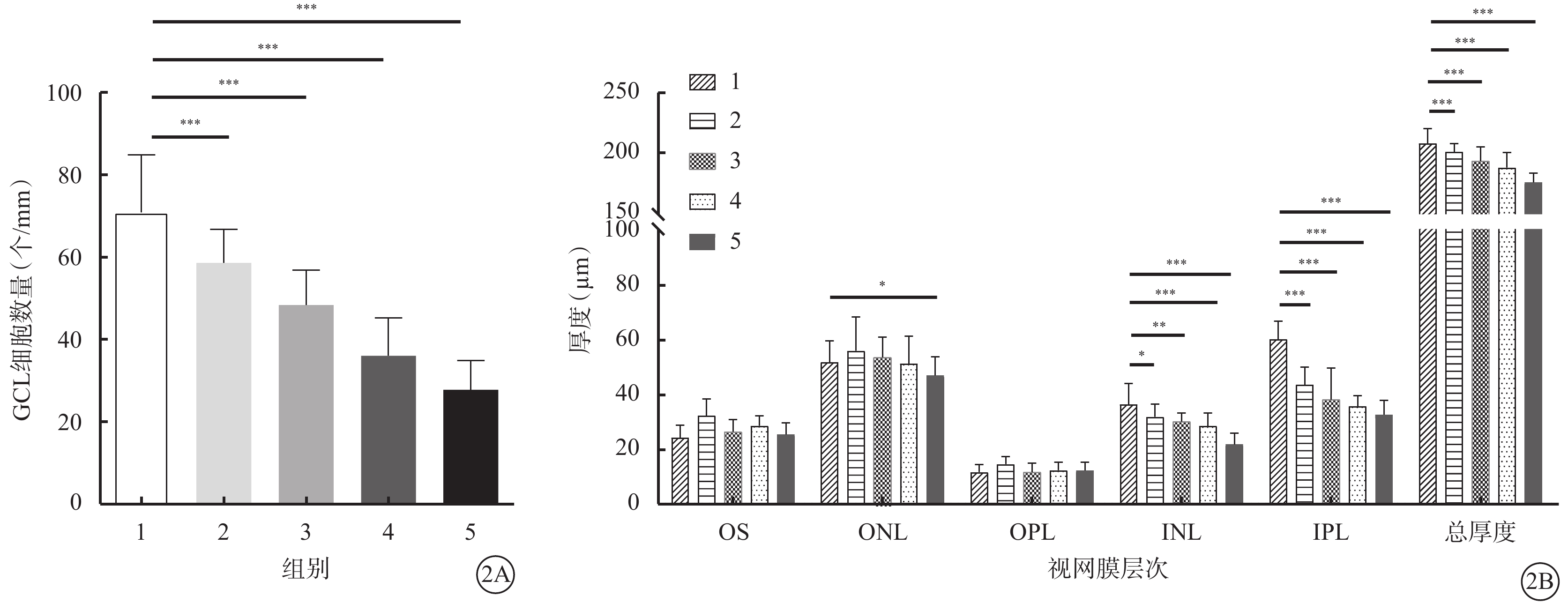 圖2
正常對照組、不同濃度NMDA組大鼠GCL細胞數量、INL和視網膜總厚度比較(n=5) NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;OS:光感受器細胞外節;ONL:外核層;OPL:外叢狀層;INL:內核層;IPL:內叢狀層;1~5分別示正常對照組、16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組。2A示GCL細胞數量比較;2B示OS、ONL、OPL、INL、IPL及視網膜總厚度比較。*P<0.05;**P<0.01;***P<0.001
圖2
正常對照組、不同濃度NMDA組大鼠GCL細胞數量、INL和視網膜總厚度比較(n=5) NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;OS:光感受器細胞外節;ONL:外核層;OPL:外叢狀層;INL:內核層;IPL:內叢狀層;1~5分別示正常對照組、16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組。2A示GCL細胞數量比較;2B示OS、ONL、OPL、INL、IPL及視網膜總厚度比較。*P<0.05;**P<0.01;***P<0.001
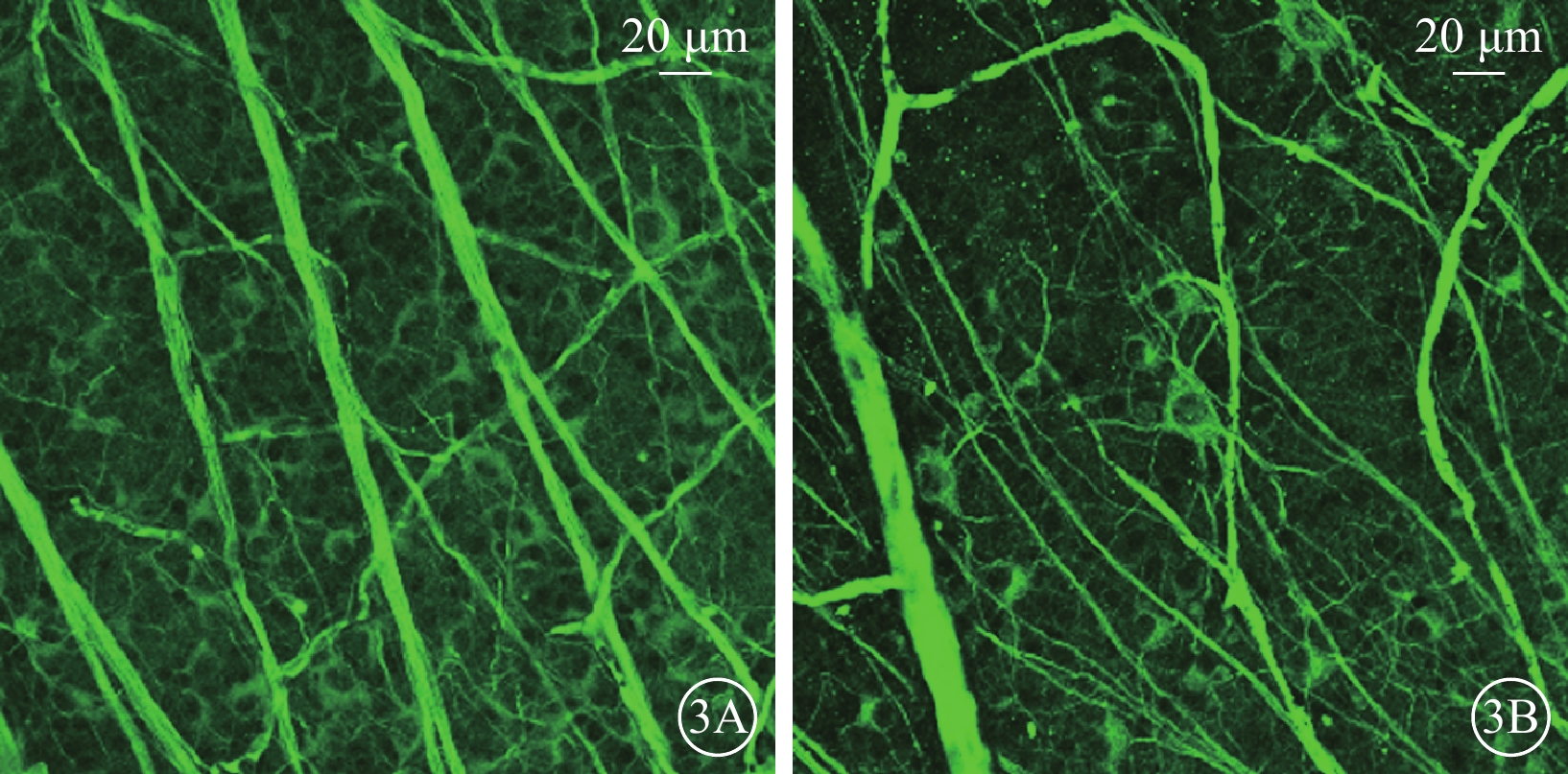
熒光顯微鏡觀察發現,正常對照組大鼠視網膜中β3微管蛋白熒光強度較強(圖3A);50.0 mmol/L NMDA組大鼠視網膜細胞β3微管蛋白熒光強度減弱(圖3B)。正常對照組、50.0 mmol/L NMDA組大鼠視網膜中β3微管蛋白染色陽性細胞數分別為(79.86±6.56)、(29.36 ±2.16)個;50.0 mmol/L NMDA組陽性細胞數顯著低于正常對照組,差異有統計學意義(t=8.67,P<0.001)(圖3)。
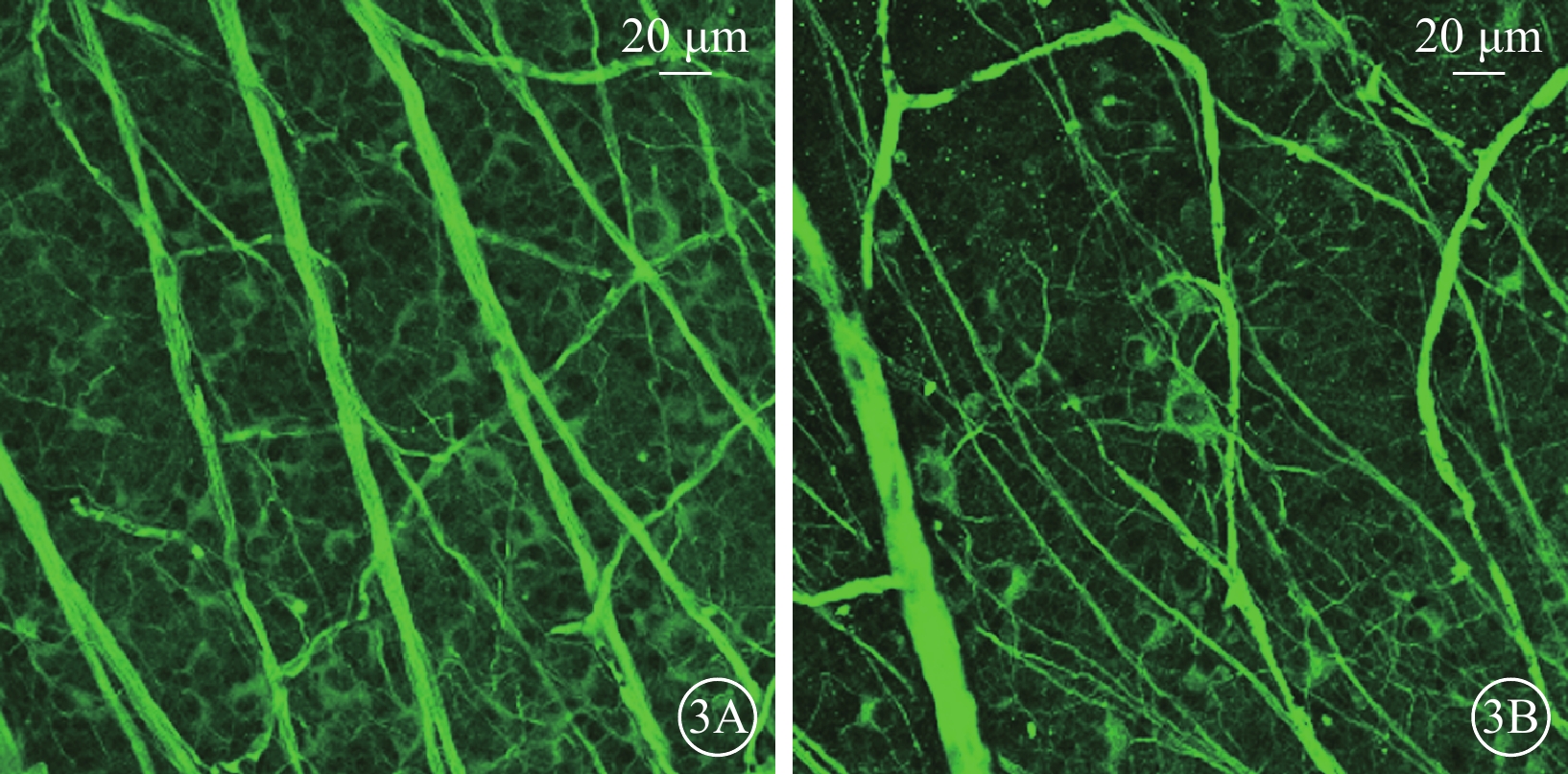 圖3
正常對照組、50.0 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視網膜熒光顯微鏡像 3A、3B分別示正常對照組、50.0 mmol/L NMDA組。與正常對照組比較,50.0 mmol/L NMDA組視網膜β3微管蛋白熒光強度減弱 標尺:20 μm
圖3
正常對照組、50.0 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視網膜熒光顯微鏡像 3A、3B分別示正常對照組、50.0 mmol/L NMDA組。與正常對照組比較,50.0 mmol/L NMDA組視網膜β3微管蛋白熒光強度減弱 標尺:20 μm
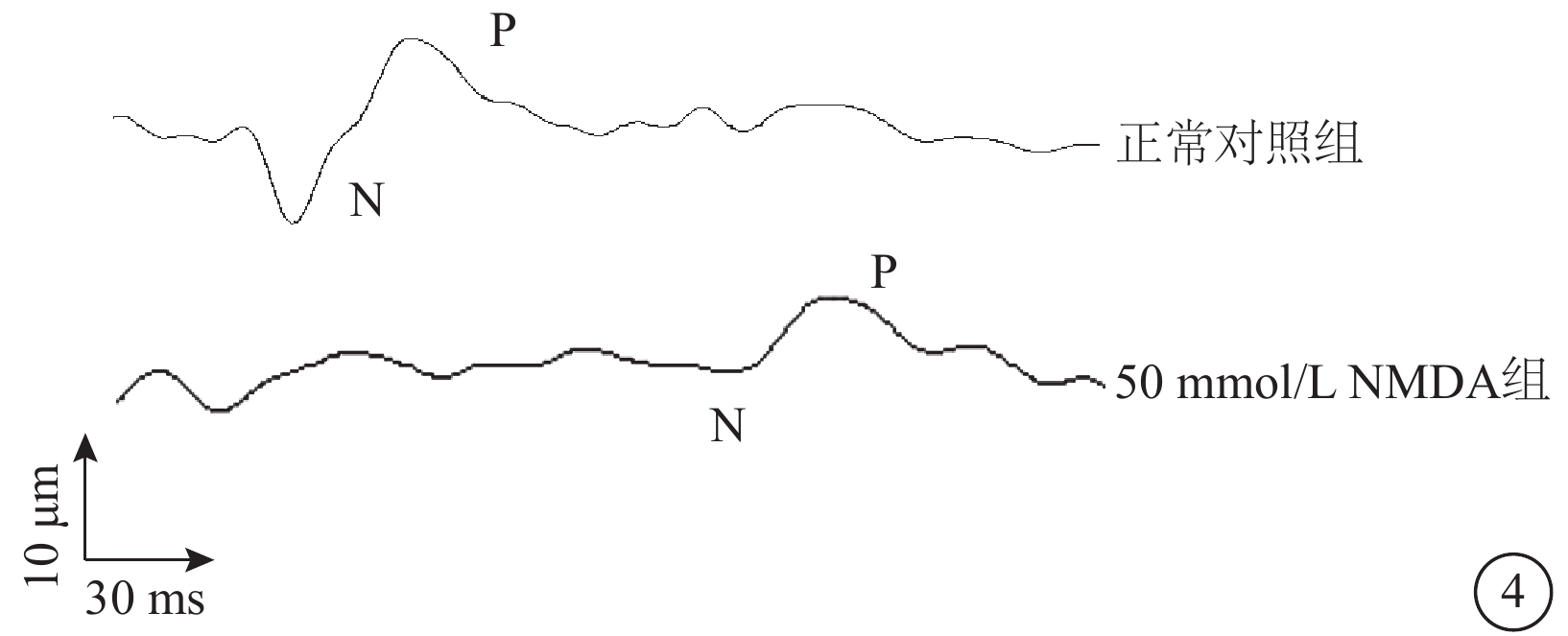
VEP檢查結果顯示,建模后7 d,正常對照組、50.0 mmol/L NMDA組P1波潛伏期分別為(117.86±6.48)、(148.46±3.78)ms;振幅分別為(42.57±2.41)、(8.68±0.63)μV。與正常對照組比較,50 mmol/L NMDA組P1波潛伏期延長,振幅顯著下降,差異均有統計學意義(t=4.37、10.50,P<0.001)(圖4)。
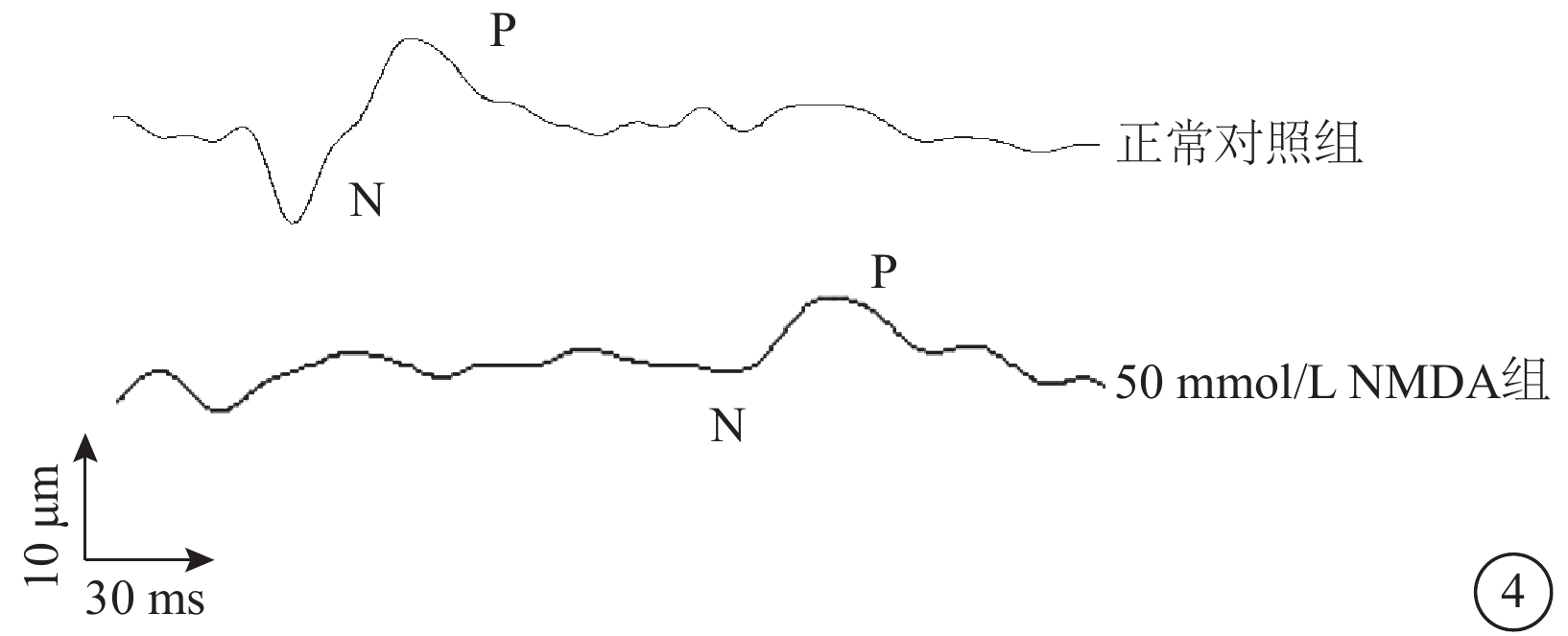 圖4
正常對照組、50 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視覺誘發電位檢查像 與正常對照組比較,50 mmol/LNMDA組P1波潛伏期延長,振幅明顯下降
圖4
正常對照組、50 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視覺誘發電位檢查像 與正常對照組比較,50 mmol/LNMDA組P1波潛伏期延長,振幅明顯下降
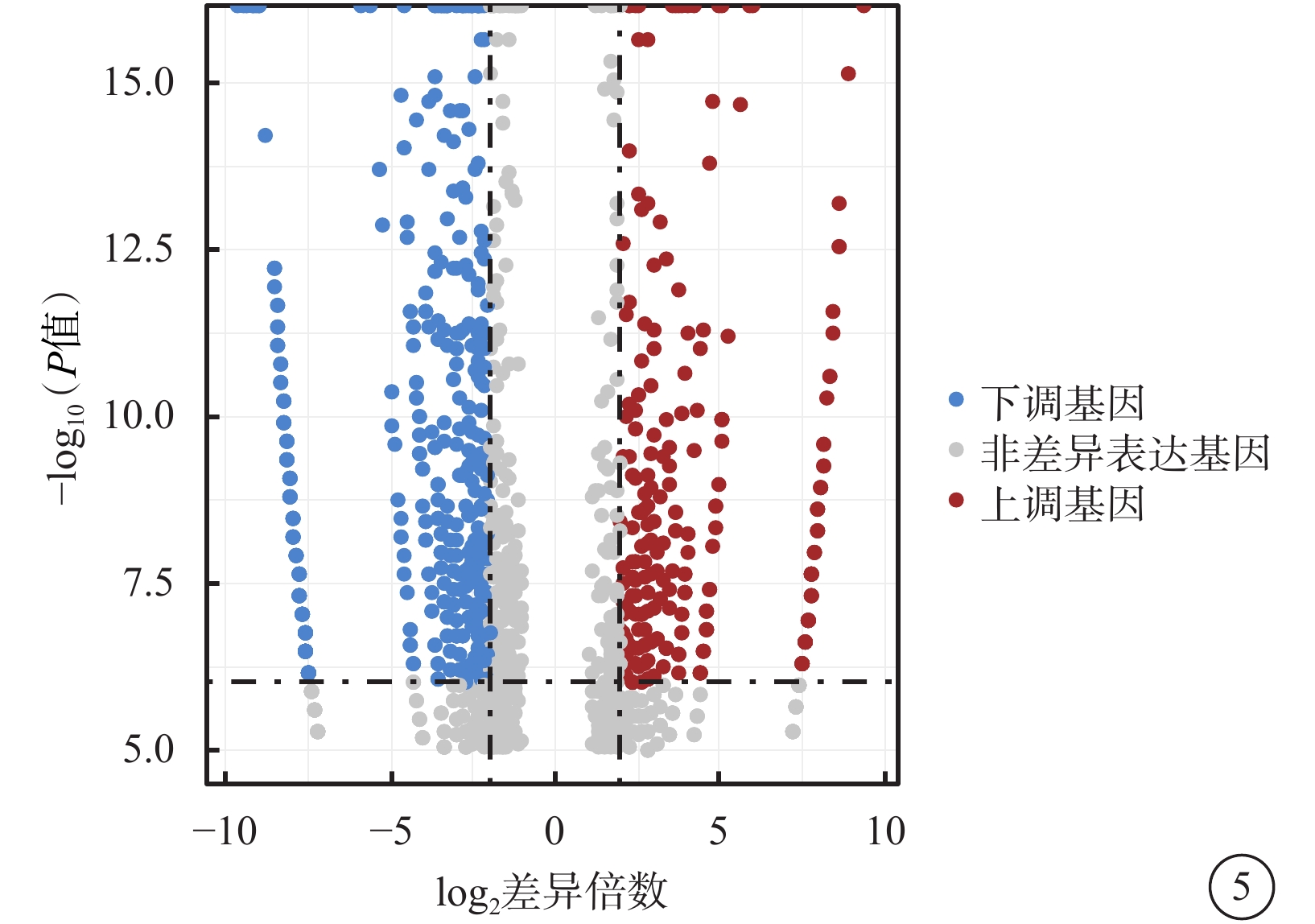
在檢測的1 181個m5C mRNA中,與正常對照組比較,50 mmol/L NMDA組篩選出差異表達m5C mRNA 576個,其中上調、下調基因分別為230、346個(圖5,表2)。
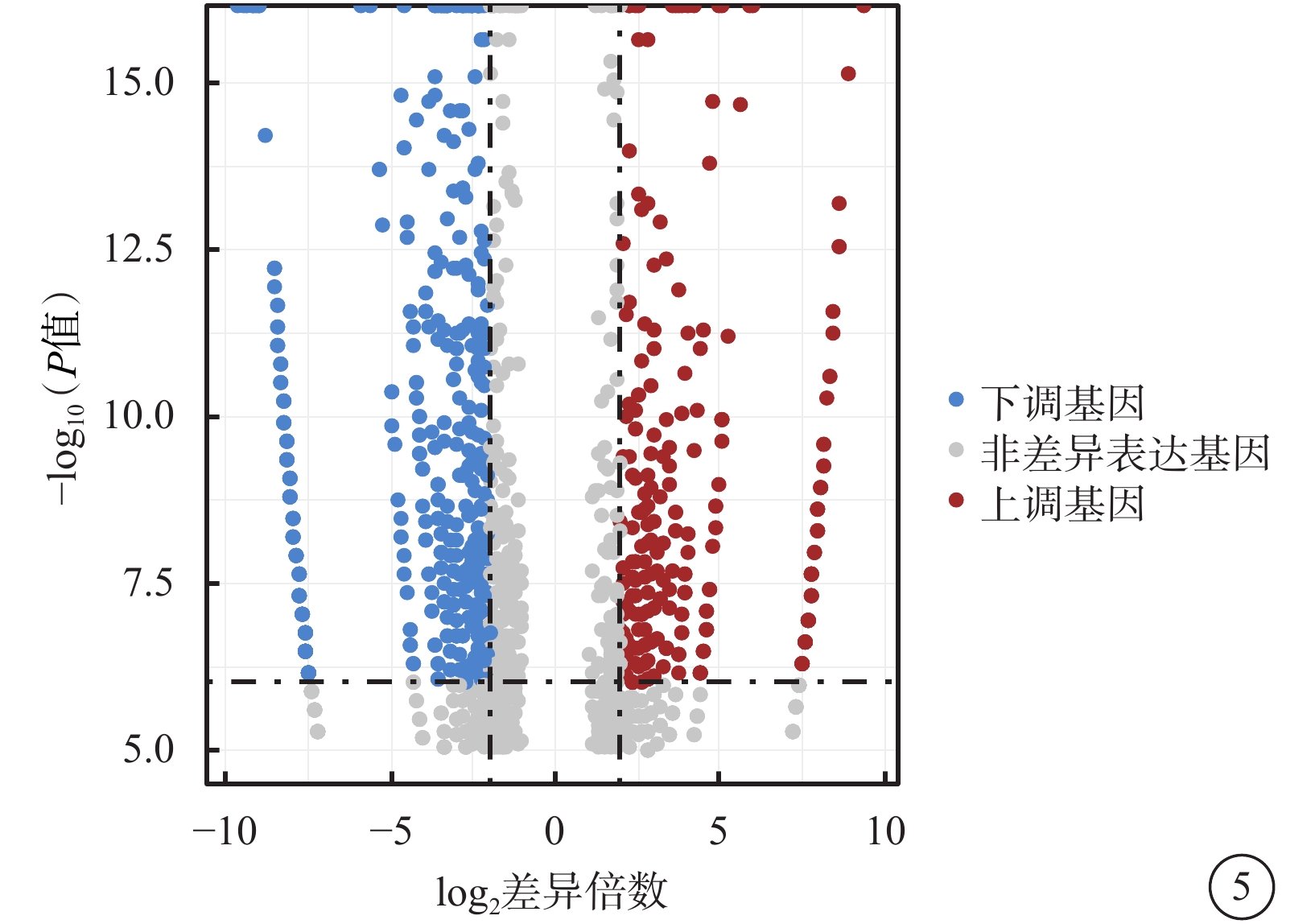 圖5
50 mmol/L N-甲基-D-天冬氨酸組差異表達基因火山圖
圖5
50 mmol/L N-甲基-D-天冬氨酸組差異表達基因火山圖
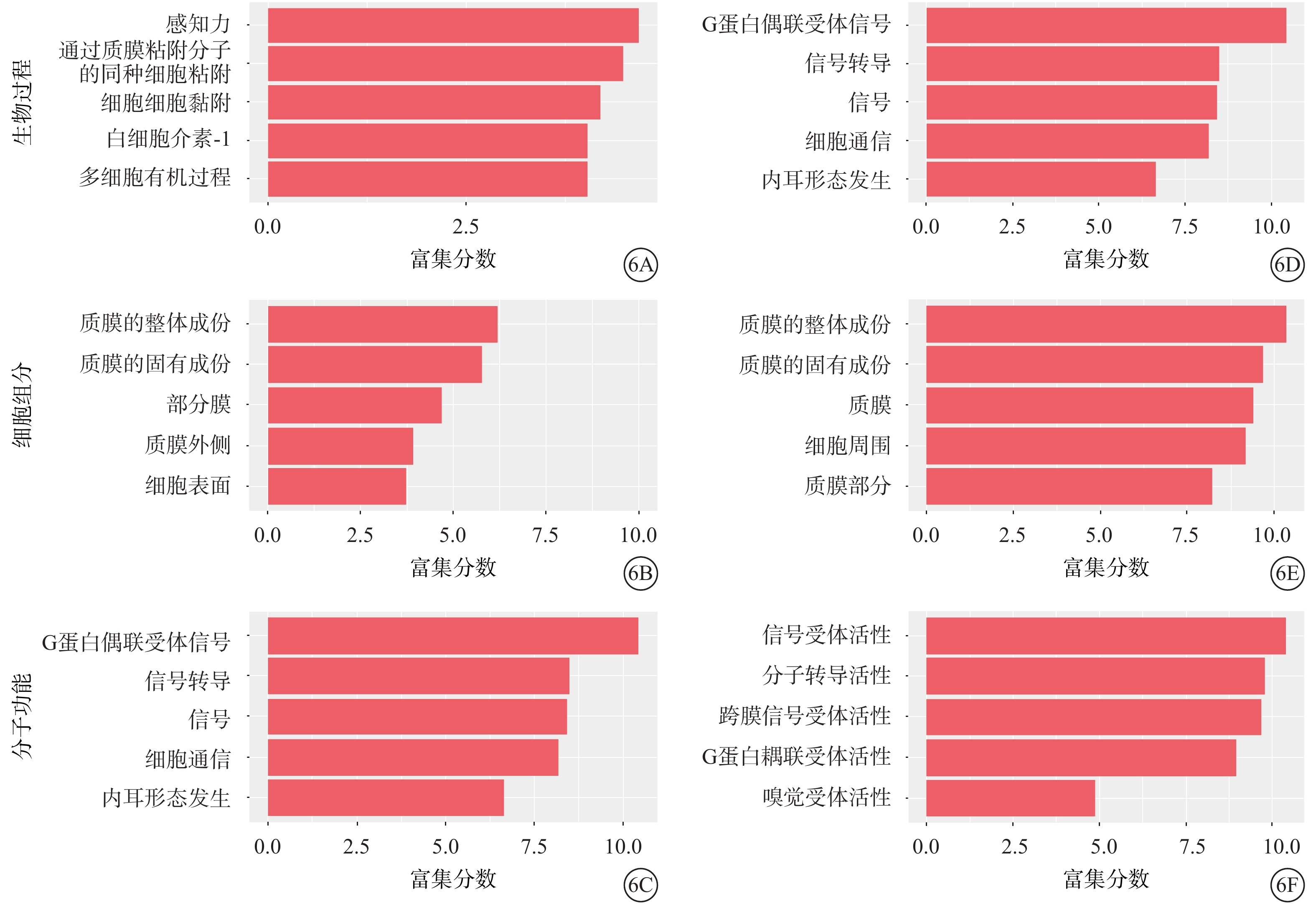
GO分析結果顯示,與正常對照組比較,50.0 mmol/L NMDA組中表達上調的m5C修飾的轉錄本主要參與感知力、通過質膜粘附分子的同種細胞粘附、細胞-細胞黏附、白細胞介素-1的產生等生物過程(圖6A);細胞膜組分包括質膜的整體成份、質膜的固有成份、部分膜、質膜外側和細胞表面(圖6B);分子功能包括信號受體活性、分子轉導活性、跨膜信號受體活性、嗅覺受體活性、G蛋白耦聯受體活性(圖6C)。表達下調的m5C修飾的轉錄本參與的生物過程主要包括G蛋白偶聯受體信號傳導途徑、信號轉導、信號以及細胞通信(圖6D);細胞膜組分包括質膜的整體成份、質膜的固有成份、質膜、細胞周圍以及質膜部分(圖6E);分子功能包括信號受體活性、分子轉導活性、跨膜信號受體活性、G蛋白耦聯受體活性以及嗅覺受體活性(圖6F)。
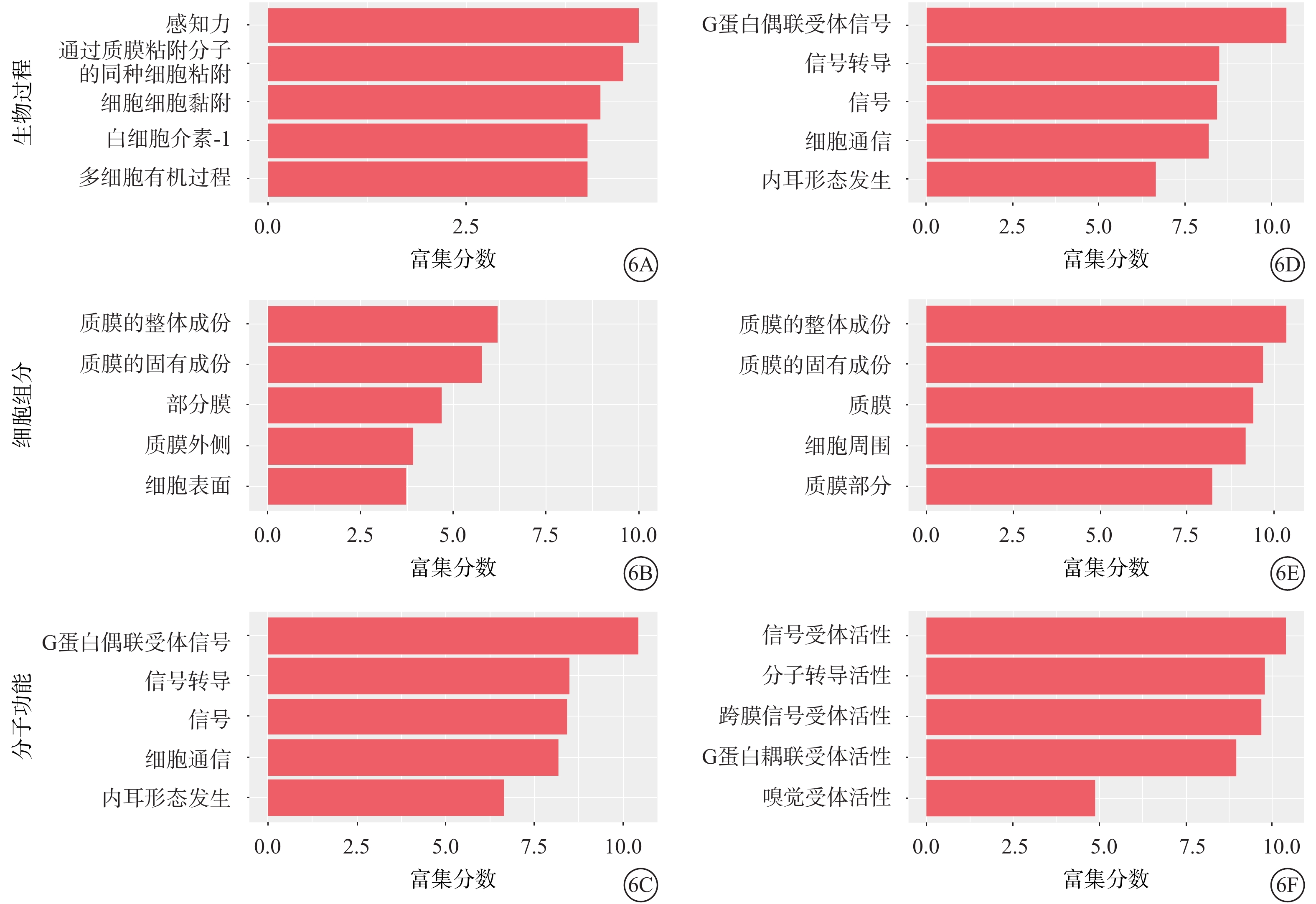 圖6
差異表達基因的基因注釋功能分類統計圖 6A~6C和6D~6F分別為上調、下調的甲基化基因
圖6
差異表達基因的基因注釋功能分類統計圖 6A~6C和6D~6F分別為上調、下調的甲基化基因
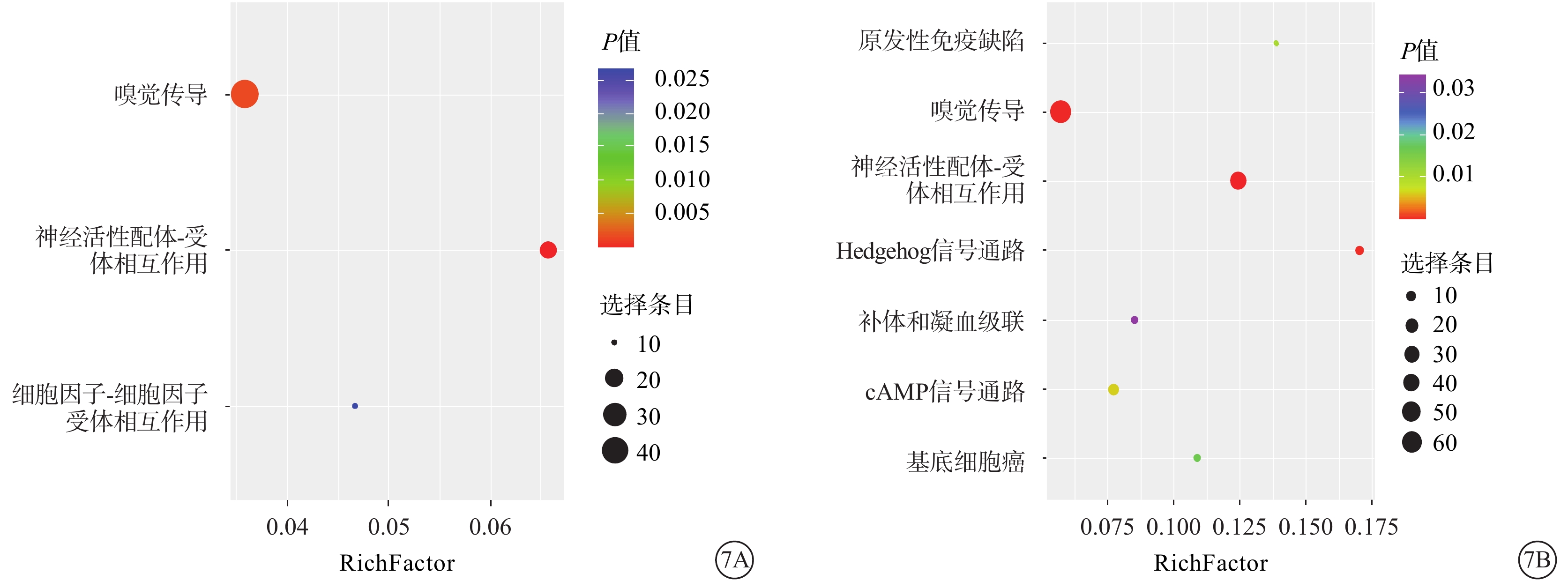
KEGG信號通路富集分析結果顯示,50.0 mmol/L NMDA組表達上調的m5C修飾的轉錄本主要富集在細胞因子-細胞因子受體的相互作用、神經活性配體-受體相互作用通路和嗅覺轉導通路(圖7A);下調的m5C修飾的轉錄本主要富集在原發性免疫缺陷通路、神經活性配體-受體相互作用通路、Hedgehog信號通路、環磷酸腺苷信號通路、基底細胞癌、補體和凝血級聯通路以及嗅覺轉導通路中(圖7B)。
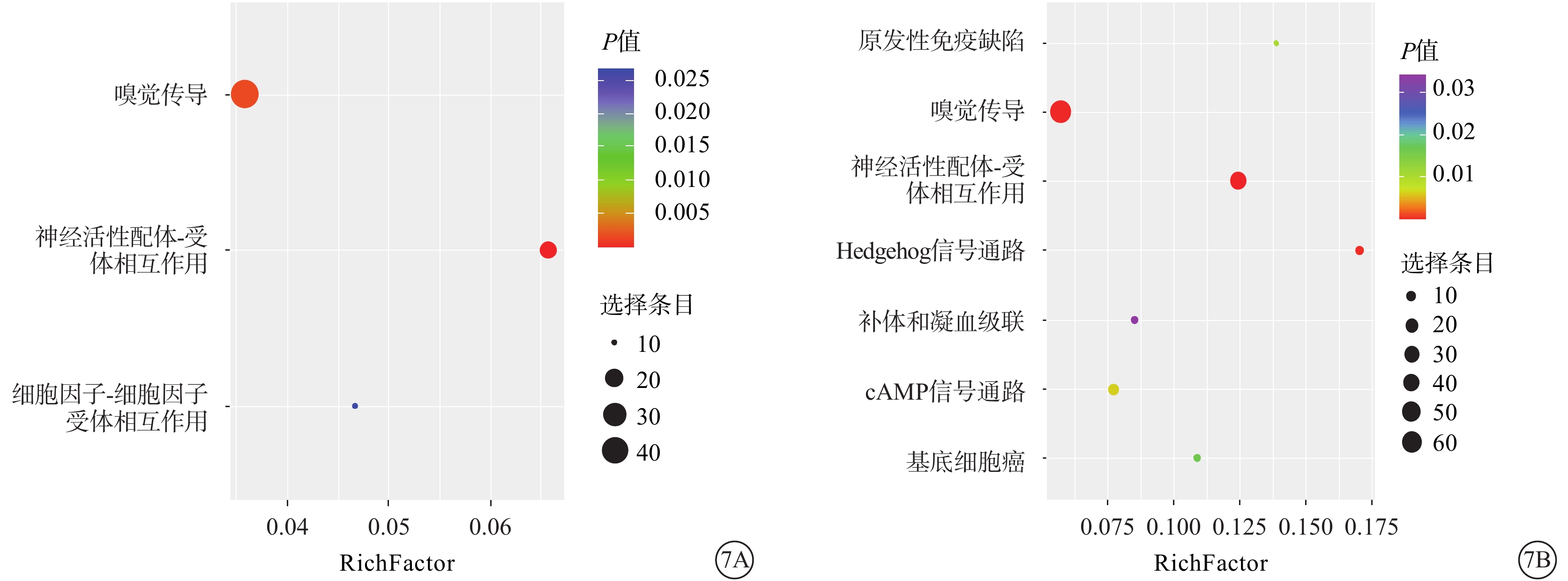 圖7
京都基因與基因組百科全書的信號通路富集分析氣泡圖 Rich Fcator:通路中差異甲基化基因數占該通路中基因總數的比例 7A、7B分別示上調、下調的甲基化基因。氣泡越大富集越顯著,顏色越深,P值越大
圖7
京都基因與基因組百科全書的信號通路富集分析氣泡圖 Rich Fcator:通路中差異甲基化基因數占該通路中基因總數的比例 7A、7B分別示上調、下調的甲基化基因。氣泡越大富集越顯著,顏色越深,P值越大
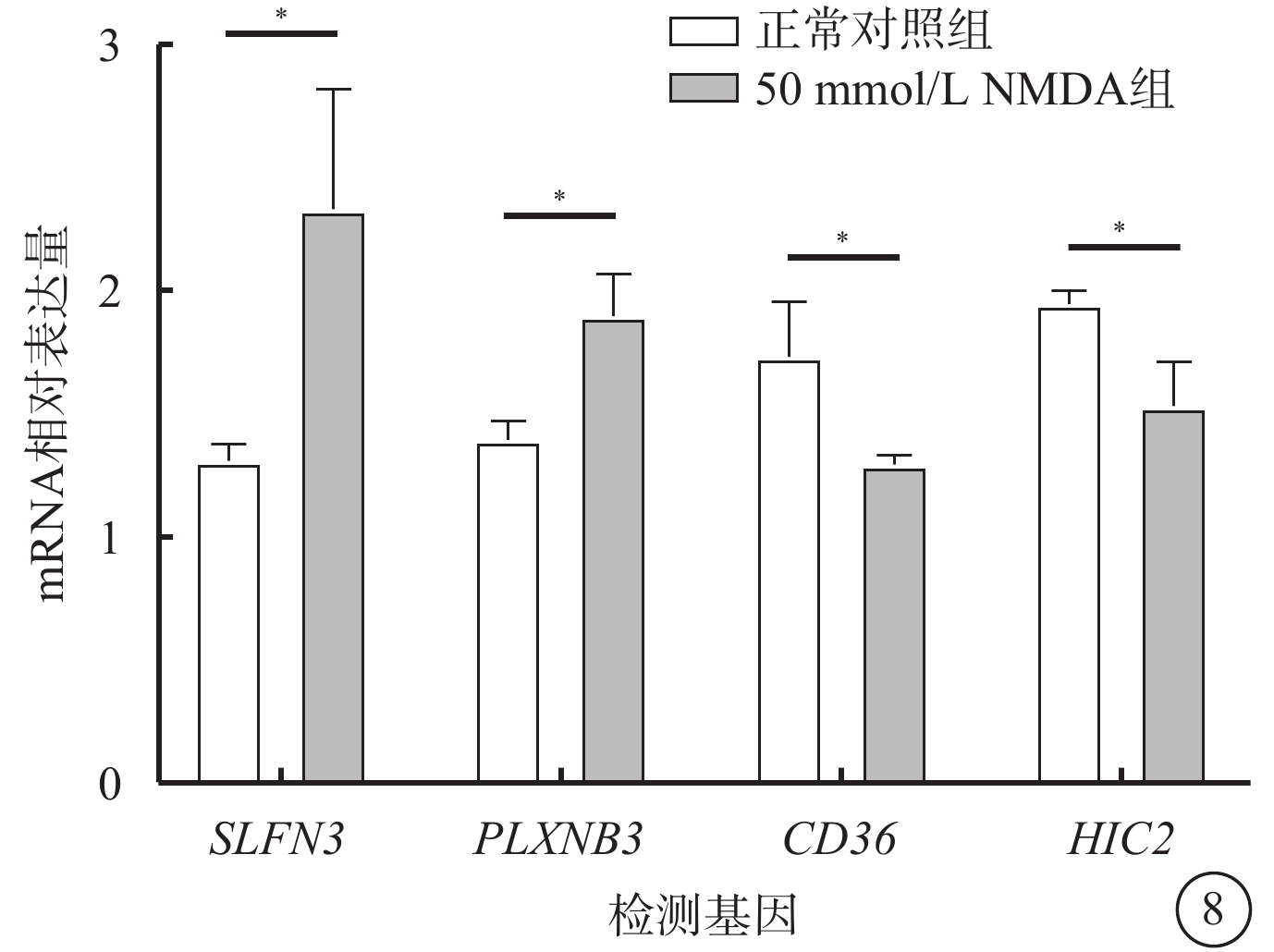
qRT-PCR檢測結果顯示,與正常對照組比較,50 mmol/L NMDA組SLFN3、 PLXNB3 mRNA相對表達量明顯升高,CD36、HIC2 mRNA相對表達量明顯降低,差異均有統計學意義(P<0.05)(圖8)。
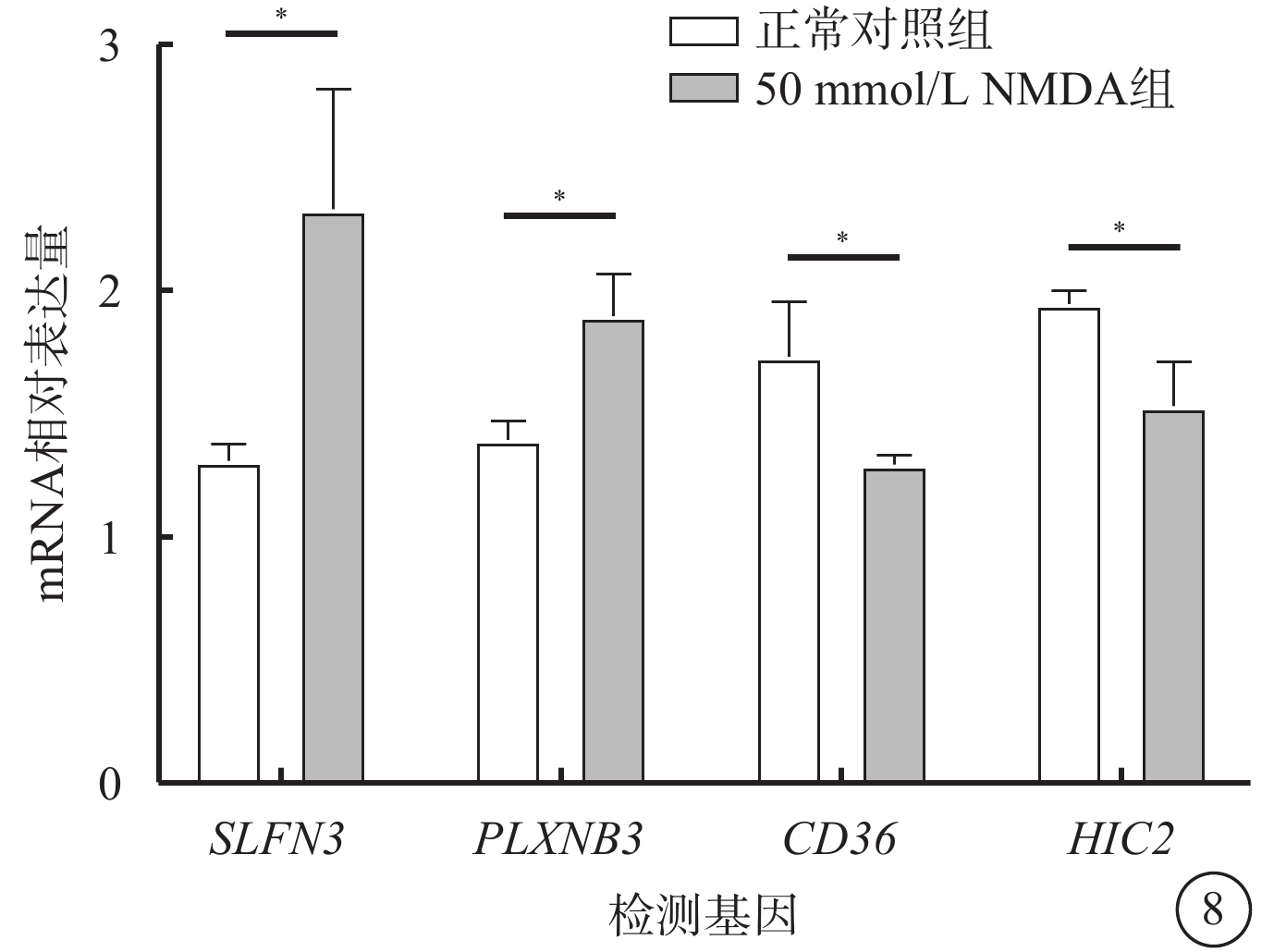 圖8
正常對照組、50 mmol/L NMDA組大鼠視網膜中SLFN3、PLXNB3、CD36、HIC2 mRNA相對表達量比較(n=8) *P<0.05
圖8
正常對照組、50 mmol/L NMDA組大鼠視網膜中SLFN3、PLXNB3、CD36、HIC2 mRNA相對表達量比較(n=8) *P<0.05
3 討論
青光眼主要特征為RGC死亡造成的不可逆視力喪失[10]。NMDA誘導的視網膜興奮性毒性是青光眼引起RGC死亡的分子機制之一。因此,保護RGC以及視網膜神經元免受NMDA誘導的興奮性毒性可能是挽救青光眼患者視力的一種可行性策略。這可以通過以下途徑實現:第一種途徑是通過拮抗NMDA受體,挽救視網膜神經元,特別是RGC的死亡。然而有學者認為完全阻斷NMDA受體可能影響視網膜的正常生理功能[11]。第二種途徑是外源性給予神經營養因子或營養物質,促進視網膜神經元的存活,但其效果缺乏臨床證據支持。因此,深入探索NMDA誘導的視網膜興奮性毒性的分子機制,可能為尋找青光眼神經保護靶點提供新的線索。
本研究結果顯示,建模后7 d,50 mmol/L NMDA組大鼠視網膜各層厚度降低,GCL細胞數量減少,視網膜鋪片中神經元個數減少,VEP測得P1波潛伏期延長、振幅顯著下降,其形態學和電生理指標均與文獻報道一致[12-13]。該結果提示NMDA誘導的視網膜興奮性毒性模型成功建立。我們進一步利用高通量MeRIP-RNA seq檢測了正常對照組和50 mmol/L NMDA組大鼠視網膜中mRNA的m5C甲基化修飾水平。測序結果表明,與正常對照組比較,50 mmol/L NMDA組視網膜的mRNA中存在1 181個m5C甲基化水平差異位點;差異表達m5C mRNA為576個,其中表達上調、下調分別為230、346個。KEGG通路分析進一步提示,NMDA組差異性表達的m5C mRNA主要富集在神經活性配體-受體相互作用通路、接受化學性刺激的嗅覺轉導通路、補體激活通路以及細胞因子-細胞因子受體的相互作用通路中。神經活性配體-受體相互作用與神經毒性有關[14]。另外,Baudouin等[15]認為補體系統的激活與RGC突觸和軸突的退行性損傷有關。
哺乳動物細胞中,多數m5C甲基化修飾位于mRNA的5’-未翻譯區和3’-未翻譯區。m5C甲基化修飾對于轉錄本功能的影響較為復雜,迄今為止,m5C在轉錄后水平上的調節被認為是其主要作用機制,即m5C修飾水平增加,mRNA穩定性增加,降解減少,mRNA編碼基因的表達上調[16-17],反之亦然。本研究驗證的4個代表性基因,包括m5C甲基化水平增高的SLFN3和PLXNB3以及m5C甲基化水平降低的CD36和HIC2,其基因表達也發生了相應的上調和下調變化,提示增加轉錄本的穩定性,調節RNA代謝可能是m5C甲基化修飾的主要作用機制之一。
另一方面,這些驗證結果為青光眼神經保護靶點的尋找提供了新線索:SLFN3的表達上調可以抑制腸道黏膜上皮細胞和腫瘤細胞的增生,促進腫瘤細胞凋亡[18-19];而PLXNB3表達上調可以促進中樞神經系統的少突膠質細胞分泌具有神經毒性的淀粉樣β蛋白[20]。這提示這兩個基因的表達上調與疾病狀態下的細胞死亡相關。CD36被認為是一種清道夫受體,其被激活后可以通過釋放大量炎癥因子而促進神經炎癥的發生[21],而其在NMDA視網膜中的表達下調,則可能是一種保護性代償機制。因此在今后的研究中,我們將嘗試調節SLFN3、PLXNB3、CD36在NMDA誘導的視網膜興奮性毒性大鼠模型中的保護作用。
本研究初步描述了NMDA誘導的視網膜興奮性毒性大鼠模型中視網膜m5C甲基化修飾的轉錄組學特征及其差異性基因表達譜,并驗證m5C修飾對基因表達的影響,提示RNA m5C修飾的表觀遺傳學調控機制可能在青光眼的發病過程發揮作用。
細胞外過量的谷氨酸可誘導N-甲基-D-天冬氨酸(NMDA)受體過度激活,造成視網膜神經節細胞(RGC)死亡和視野進行性縮窄,這些病理改變在青光眼等致盲性眼病中已得到廣泛研究并冠名為“視網膜興奮性毒性”[1]。但NMDA誘導的視網膜興奮性毒性的分子機制,特別是RNA甲基化修飾的表觀遺傳學調控機制,仍屬未知。RNA甲基化修飾是指在RNA分子不同位置上的甲基化修飾現象,包括6-甲基腺嘌呤(m6A)、5-甲基胞嘧啶(m5C)等形式[2-3]。文獻報道,RNA甲基化修飾在眼部疾病中主要表現為m6A甲基化的分布與調控異常[4-6];但在人轉錄組中,20%~30%的mRNA帶有m5C的甲基化修飾[7],提示m5C甲基化修飾在生理和病理條件下的調控作用不容忽視。目前m5C甲基化修飾在視網膜興奮性毒性中的致病機制國內外文獻均鮮見報道。因此,本研究在NMDA誘導的大鼠視網膜興奮性毒性模型中,利用高通量m5C甲基化RNA測序(MeRIP-RNA seq)檢測視網膜中轉錄本m5C甲基化的修飾水平,并輔以生物信息學分析,并初步驗證m5C甲基化修飾對基因表達的影響。現將結果報道如下。
1 材料和方法
1.1 材料
實驗動物。Sprague Dawley(SD)雄性大鼠65只,7~8周齡,體重200~250 g;分兩批購自北京維通利華公司,飼養于天津醫科大學眼科醫院無特定病原體級標準(12 h明/12 h暗循環)動物房。本研究經天津醫科大學動物倫理委員會審核通過(動物倫理批號:SYXK 2009-0001)。所有實驗操作均符合國家衛生研究院實驗室動物護理和使用指南;實驗動物使用遵循視覺與眼科研究協會有關眼科與視覺科學研究中動物使用的相關規定。
主要試劑及儀器。NMDA(美國Sigma公司);大鼠單克隆β3-微管蛋白抗體、兔抗大鼠二抗(美國Abcam公司);抗淬滅封片劑(美國Vectashield公司);RNA提取試劑盒(美國EZBioscience公司);氧化鋯研磨珠高速組織研磨儀(武漢塞維爾生物科技有限公司);共聚焦顯微鏡(德國Zeiss公司)。
1.2 確立建模的NMDA最佳玻璃體腔注射濃度
篩選建立視網膜興奮性毒性模型時NMDA的最佳玻璃體腔注射濃度。首批25只SD大鼠適應環境7 d后,隨機分為正常對照組以及NMDA 16.7、33.3、50.0、66.7 mmol/L組(每組5只)。NMDA溶解于質量分數為0.9%的生理鹽水中,濾膜(孔徑0.2 μm)過濾,制備16.7、33.3、50.0、66.7 mmol/L無菌NMDA溶液。大鼠按體重腹腔注射40 mg/kg戊巴比妥鈉全身麻醉,鹽酸奧布卡因滴眼液眼表麻醉。不同濃度NMDA組大鼠右眼(模型眼)玻璃體腔注射相應濃度3 μl NMDA,正常對照組大鼠右眼玻璃體腔注射等體積生理鹽水。已剔除虹膜出血、晶狀體受損、穿刺及拔針過程中漏液過多以及注射后出現眼部感染的大鼠。
蘇木素-伊紅(HE)染色觀察正常對照組、不同濃度NMDA組大鼠的視網膜整體結構。建模后7 d,大鼠腹腔注射過量戊巴比妥鈉深度麻醉處死。摘除眼球,置于酸性固定液中固定。常規石蠟包埋,于視神經附近沿眼球縱向進行切片,每個眼球在相似部位連續切片5張,3 μm/張。組織切片脫蠟、脫水,HE染色,光學顯微鏡下觀察拍照。采用CellSense軟件測量視網膜各層厚度,Photoshop CS6計數視網膜神經節細胞層(GCL)中細胞數量。所有操作均采用雙盲原則。依據文獻[8-9],原發性開角型青光眼患者臨床常規檢測中發現視野缺損時,已有50%的節細胞丟失;而本研究實驗結果顯示,50 mmol/L NMDA經玻璃體腔注射7 d后,大鼠GCL細胞數量減少到正常對照組的50.09%。為使動物模型再現臨床病理改變,選取玻璃體腔注射50 mmol/L NMDA,建立視網膜興奮性毒性大鼠模型。
1.3 實驗方法
第二批SD大鼠40只,隨機分為正常對照組、50 mmol/L NMDA組,每組各20只。
視覺誘發電位(VEP)檢測大鼠視神經傳導功能。建模后7 d,過夜暗適應后,正常對照組、50 mmol/L NMDA組大鼠,按體重腹腔注射40 mg/kg戊巴比妥鈉全身麻醉,右眼(模型眼)復方托吡卡胺散瞳。弱紅光下,大鼠以俯臥位固定于實驗臺上,將針狀鉑電極置于大鼠角膜,參考電極置于枕骨粗隆,地電極插入尾部皮下,按視覺電生理記錄儀說明書,給予大鼠30.0 cd·s/m2白色閃光刺激。右眼檢查時,左眼不透光眼罩完全遮蓋。記錄大鼠全視野暗適應VEP參數,包括P1波振幅和P1波潛伏期。P1波潛伏期為起點至P1波波峰時間,P1波振幅為起點至P1波峰距離。連續測量3次,取平均值。
視網膜鋪片免疫熒光染色檢測β3微管蛋白免疫熒光染色陽性細胞個數。選取正常對照組、50 mmol/L NMDA組大鼠各9只過量麻醉處死,摘除眼球,4%多聚甲醛(PFA)固定40 min,手術顯微鏡下剝離視網膜,置于含4% PFA的24孔板中,4 ℃固定過夜。磷酸鹽緩沖液(PBS)洗2次,每孔加入1 ml通透液,4 ℃孵育過夜。PBlec緩沖液洗3次。加入β3微管蛋白抗體(鼠源單抗體,1∶1 000,PBlec緩沖液稀釋),室溫孵育過夜。PBS洗3次,加入異硫氰酸熒光素(FITC)標記的二抗(兔抗鼠,1∶500,PBlec緩沖液稀釋),PBS洗3次。將僅加FITC標記的熒光二抗染色設為陰性對照。將視網膜呈放射狀切成3~4瓣,GCL向上平鋪于載玻片上,滴加少量含4',6-二脒基-2-苯基吲哚的抗熒光衰退封片劑封片。共聚焦顯微鏡下觀察拍照。將熒光信號強度高于非特異性背景,且呈現細胞形態的胞漿綠色熒光染色視為β3微管蛋白染色陽性,應用Photoshop CS6軟件計數β3微管蛋白染色陽性的細胞數。
MeRIP-RNA seq及生物信息學分析。建模后7 d,正常對照組和50.0 mmol/L NMDA組各收集3只眼,冰上迅速剝離視網膜,置于EP管中,液氮速凍,應用MeRIP-RNA測序進行m5C表觀轉錄組學分析,由上海云序生物科技有限公司完成。采用GeneJET RNA提取試劑盒提取各組標本總RNA,GenSeq m5C RNA IP試劑盒對m5C修飾的RNA進行免疫沉淀,富集m5C修飾的轉錄本。應用NEBNext Ultra II Directional RNA Library Prep試劑盒對免疫沉淀后的m5C RNA樣本進行RNA測序文庫的構建與質檢。使用Illumina HiSeq 4000測序儀測序、堿基識別和質控,產生原始測序數據,經Q30質控、去接頭,獲得高質量數據。采用Hisat2軟件(v2.0.4)將全部樣本的高質量數據匹配到參考基因組上。應用MACS軟件識別每個樣本中的甲基化基因,diffReps軟件進行差異甲基化基因的識別,以正常對照組和50.0 mmol/L NMDA組之間的差異倍數≥2和P≤0.000 1為標準,滿足此二項條件者,鑒定為差異甲基化基因;再應用公司自有程序篩選位于mRNA外顯子上的m5C甲基化峰,進行相應注釋。最后,對差異甲基化的編碼基因進行基因注釋(GO)和京都基因與基因組百科全書(KEGG)分析。
根據m5C MeRIP-RNA測序結果,選取NMDA處理后,m5C修飾水平增高(SLFN3
總RNA的提取和逆轉錄。建模后7 d,選取正常對照組、50 mmol/L NMDA組大鼠各8只,過量麻醉處死。收集視網膜,液氮速凍,-80 ℃冰箱儲存。每個視網膜置于1個1.5 ml EP管中,分別加入直徑4、3 mm的氧化鋯研磨珠1、2,500 μl裂解液作用,于高速組織研磨器中充分研磨。研磨后液體移至另一1.5 ml EP管,按Universal RNA Purification Kit試劑盒說明書,提取視網膜組織中總RNA。應用Nanodrop 2000微量分光光度計測定所得總RNA的濃度和純度。取1 μg總RNA,按照RevertAid First Strand cDNA Synthesis Kit說明書,用隨機六聚體引物,行逆轉錄,制備cDNA。
實時定量聚合酶鏈反應(qRT-PCR)檢測正常對照組、50 mmol/L NMDA組視網膜中SLFN3、PLXNB3、CD36、HIC2 mRNA相對表達量。PubMed中查詢大鼠4個目標基因(SLFN3、PLXNB3、CD36、HIC2)以及內參基因GAPDH的序列,應用Primer Express 3.0軟件設計引物序列(表1),由深圳華大基因股份有限公司合成。以4 μl cDNA為模板,加入基因的正向、反向引物各1.5 μl,以及12.5 μl EvaGreen 2×qPCR Master Mix,再加入無核酸酶的水,補足25 μl反應體系,在96孔板中進行qRT-PCR擴增。反應條件為:50 ℃孵育2 min,95 ℃變性10 min;95 ℃變性15 s,60 ℃退火和延伸1 min,40個循環;95 ℃變性15 s,60 ℃退火15 s,95 ℃反應15 s。采用2?△△Ct法計算目的基因mRNA相對表達情況。
1.4 統計學方法
采用GraphPad Prism 8軟件進行統計學分析。各組數據資料經Shapiro-Wilk檢驗呈正態分布,經Levene檢驗證實方差齊,并以均數±標準差(±s)表示。正常對照組、不同濃度NMDA組大鼠視網膜各層厚度、GCL細胞數量比較采用單因素方差分析,組間多重比較采用Tukey post hoc檢驗;正常對照組與50 mmol/L NMDA組間β3-微管蛋白染色陽性細胞數、VEP P1波振幅和潛伏期、mRNA相對表達量比較采用雙側非配對t檢驗。P<0.05為差異有統計學意義。
2 結果
光學顯微鏡觀察發現,建模后7 d,正常對照組大鼠視網膜各層清晰,組織形態完整,細胞排列規則(圖1A)。16.7 mmol/L NMDA組大鼠視網膜GCL細胞空泡變性(圖1B);33.3 mmol/L NMDA組大鼠視網膜GCL細胞數量相對減少,排列紊亂,細胞內空泡變性(圖1C);50 mmol/L NMDA組大鼠視網膜GCL細胞數量明顯減少且伴視網膜水腫(圖1D);66.7 mmol/L NMDA組大鼠視網膜GCL細胞數量變少,核固縮,內核層變薄且紊亂,視網膜總厚度變薄(圖1E)。
圖1
正常對照組、不同濃度NMDA組大鼠視網膜組織光學顯微鏡像 NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;IPL:內叢狀層;INL:內核層;OPL:外叢狀層;ONL:外核層;OS:光感受器細胞外節。1A示正常對照組,視網膜各層清晰,組織形態完整,細胞排列規則;1B示16.7 mmol/L NMDA組,GCL細胞空泡變性;1C示33.3 mmol/L NMDA組,GCL細胞數量相對減少,排列紊亂,細胞內空泡變性;1D示50.0 mmol/L NMDA組,GCL細胞數量明顯減少且伴視網膜水腫;1E示66.7 mmol/L NMDA組,GCL細胞數量變少,核固縮,內核層變薄且紊亂 蘇木精-伊紅染色 標尺:20 μm
正常對照組、不同濃度NMDA組大鼠視網膜GCL細胞數量及內核層、內叢狀層、外核層、視網膜總厚度比較,差異均有統計學意義(P<0.05);外叢狀層、光感受器細胞外節厚度比較,差異無統計學意義(P>0.05)(圖2)。與正常對照組比較,16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組GCL細胞數量明顯減少,其中16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組GCL細胞數量分別為正常對照組的81.57%、67.18%、50.09%、38.64%,差異均有統計學意義(P<0.001);不同濃度NMDA組內核層厚度、視網膜總厚度均明顯降低,差異均有統計學意義(P<0.001)。
圖2
正常對照組、不同濃度NMDA組大鼠GCL細胞數量、INL和視網膜總厚度比較(n=5) NMDA:N-甲基-D-天冬氨酸;GCL:神經節細胞層;OS:光感受器細胞外節;ONL:外核層;OPL:外叢狀層;INL:內核層;IPL:內叢狀層;1~5分別示正常對照組、16.7 mmol/L NMDA組、33.3 mmol/L NMDA組、50.0 mmol/L NMDA組、66.7 mmol/L NMDA組。2A示GCL細胞數量比較;2B示OS、ONL、OPL、INL、IPL及視網膜總厚度比較。*P<0.05;**P<0.01;***P<0.001
熒光顯微鏡觀察發現,正常對照組大鼠視網膜中β3微管蛋白熒光強度較強(圖3A);50.0 mmol/L NMDA組大鼠視網膜細胞β3微管蛋白熒光強度減弱(圖3B)。正常對照組、50.0 mmol/L NMDA組大鼠視網膜中β3微管蛋白染色陽性細胞數分別為(79.86±6.56)、(29.36 ±2.16)個;50.0 mmol/L NMDA組陽性細胞數顯著低于正常對照組,差異有統計學意義(t=8.67,P<0.001)(圖3)。
圖3
正常對照組、50.0 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視網膜熒光顯微鏡像 3A、3B分別示正常對照組、50.0 mmol/L NMDA組。與正常對照組比較,50.0 mmol/L NMDA組視網膜β3微管蛋白熒光強度減弱 標尺:20 μm
VEP檢查結果顯示,建模后7 d,正常對照組、50.0 mmol/L NMDA組P1波潛伏期分別為(117.86±6.48)、(148.46±3.78)ms;振幅分別為(42.57±2.41)、(8.68±0.63)μV。與正常對照組比較,50 mmol/L NMDA組P1波潛伏期延長,振幅顯著下降,差異均有統計學意義(t=4.37、10.50,P<0.001)(圖4)。
圖4
正常對照組、50 mmol/L N-甲基-D-天冬氨酸(NMDA)組大鼠視覺誘發電位檢查像 與正常對照組比較,50 mmol/LNMDA組P1波潛伏期延長,振幅明顯下降
在檢測的1 181個m5C mRNA中,與正常對照組比較,50 mmol/L NMDA組篩選出差異表達m5C mRNA 576個,其中上調、下調基因分別為230、346個(圖5,表2)。
圖5
50 mmol/L N-甲基-D-天冬氨酸組差異表達基因火山圖
GO分析結果顯示,與正常對照組比較,50.0 mmol/L NMDA組中表達上調的m5C修飾的轉錄本主要參與感知力、通過質膜粘附分子的同種細胞粘附、細胞-細胞黏附、白細胞介素-1的產生等生物過程(圖6A);細胞膜組分包括質膜的整體成份、質膜的固有成份、部分膜、質膜外側和細胞表面(圖6B);分子功能包括信號受體活性、分子轉導活性、跨膜信號受體活性、嗅覺受體活性、G蛋白耦聯受體活性(圖6C)。表達下調的m5C修飾的轉錄本參與的生物過程主要包括G蛋白偶聯受體信號傳導途徑、信號轉導、信號以及細胞通信(圖6D);細胞膜組分包括質膜的整體成份、質膜的固有成份、質膜、細胞周圍以及質膜部分(圖6E);分子功能包括信號受體活性、分子轉導活性、跨膜信號受體活性、G蛋白耦聯受體活性以及嗅覺受體活性(圖6F)。
圖6
差異表達基因的基因注釋功能分類統計圖 6A~6C和6D~6F分別為上調、下調的甲基化基因
KEGG信號通路富集分析結果顯示,50.0 mmol/L NMDA組表達上調的m5C修飾的轉錄本主要富集在細胞因子-細胞因子受體的相互作用、神經活性配體-受體相互作用通路和嗅覺轉導通路(圖7A);下調的m5C修飾的轉錄本主要富集在原發性免疫缺陷通路、神經活性配體-受體相互作用通路、Hedgehog信號通路、環磷酸腺苷信號通路、基底細胞癌、補體和凝血級聯通路以及嗅覺轉導通路中(圖7B)。
圖7
京都基因與基因組百科全書的信號通路富集分析氣泡圖 Rich Fcator:通路中差異甲基化基因數占該通路中基因總數的比例 7A、7B分別示上調、下調的甲基化基因。氣泡越大富集越顯著,顏色越深,P值越大
qRT-PCR檢測結果顯示,與正常對照組比較,50 mmol/L NMDA組SLFN3、 PLXNB3 mRNA相對表達量明顯升高,CD36、HIC2 mRNA相對表達量明顯降低,差異均有統計學意義(P<0.05)(圖8)。
圖8
正常對照組、50 mmol/L NMDA組大鼠視網膜中SLFN3、PLXNB3、CD36、HIC2 mRNA相對表達量比較(n=8) *P<0.05
3 討論
青光眼主要特征為RGC死亡造成的不可逆視力喪失[10]。NMDA誘導的視網膜興奮性毒性是青光眼引起RGC死亡的分子機制之一。因此,保護RGC以及視網膜神經元免受NMDA誘導的興奮性毒性可能是挽救青光眼患者視力的一種可行性策略。這可以通過以下途徑實現:第一種途徑是通過拮抗NMDA受體,挽救視網膜神經元,特別是RGC的死亡。然而有學者認為完全阻斷NMDA受體可能影響視網膜的正常生理功能[11]。第二種途徑是外源性給予神經營養因子或營養物質,促進視網膜神經元的存活,但其效果缺乏臨床證據支持。因此,深入探索NMDA誘導的視網膜興奮性毒性的分子機制,可能為尋找青光眼神經保護靶點提供新的線索。
本研究結果顯示,建模后7 d,50 mmol/L NMDA組大鼠視網膜各層厚度降低,GCL細胞數量減少,視網膜鋪片中神經元個數減少,VEP測得P1波潛伏期延長、振幅顯著下降,其形態學和電生理指標均與文獻報道一致[12-13]。該結果提示NMDA誘導的視網膜興奮性毒性模型成功建立。我們進一步利用高通量MeRIP-RNA seq檢測了正常對照組和50 mmol/L NMDA組大鼠視網膜中mRNA的m5C甲基化修飾水平。測序結果表明,與正常對照組比較,50 mmol/L NMDA組視網膜的mRNA中存在1 181個m5C甲基化水平差異位點;差異表達m5C mRNA為576個,其中表達上調、下調分別為230、346個。KEGG通路分析進一步提示,NMDA組差異性表達的m5C mRNA主要富集在神經活性配體-受體相互作用通路、接受化學性刺激的嗅覺轉導通路、補體激活通路以及細胞因子-細胞因子受體的相互作用通路中。神經活性配體-受體相互作用與神經毒性有關[14]。另外,Baudouin等[15]認為補體系統的激活與RGC突觸和軸突的退行性損傷有關。
哺乳動物細胞中,多數m5C甲基化修飾位于mRNA的5’-未翻譯區和3’-未翻譯區。m5C甲基化修飾對于轉錄本功能的影響較為復雜,迄今為止,m5C在轉錄后水平上的調節被認為是其主要作用機制,即m5C修飾水平增加,mRNA穩定性增加,降解減少,mRNA編碼基因的表達上調[16-17],反之亦然。本研究驗證的4個代表性基因,包括m5C甲基化水平增高的SLFN3和PLXNB3以及m5C甲基化水平降低的CD36和HIC2,其基因表達也發生了相應的上調和下調變化,提示增加轉錄本的穩定性,調節RNA代謝可能是m5C甲基化修飾的主要作用機制之一。
另一方面,這些驗證結果為青光眼神經保護靶點的尋找提供了新線索:SLFN3的表達上調可以抑制腸道黏膜上皮細胞和腫瘤細胞的增生,促進腫瘤細胞凋亡[18-19];而PLXNB3表達上調可以促進中樞神經系統的少突膠質細胞分泌具有神經毒性的淀粉樣β蛋白[20]。這提示這兩個基因的表達上調與疾病狀態下的細胞死亡相關。CD36被認為是一種清道夫受體,其被激活后可以通過釋放大量炎癥因子而促進神經炎癥的發生[21],而其在NMDA視網膜中的表達下調,則可能是一種保護性代償機制。因此在今后的研究中,我們將嘗試調節SLFN3、PLXNB3、CD36在NMDA誘導的視網膜興奮性毒性大鼠模型中的保護作用。
本研究初步描述了NMDA誘導的視網膜興奮性毒性大鼠模型中視網膜m5C甲基化修飾的轉錄組學特征及其差異性基因表達譜,并驗證m5C修飾對基因表達的影響,提示RNA m5C修飾的表觀遺傳學調控機制可能在青光眼的發病過程發揮作用。