引用本文: 張琦穎, 申璐, 楊文昌, 代敏, 鄭宇祥, 曾文一, 胡豫, 李冬莉, 羅艷紅, 袁玲. Sorsby眼底營養不良一家系2例. 中華眼底病雜志, 2023, 39(7): 588-590. doi: 10.3760/cma.j.cn511434-20220328-00173 復制
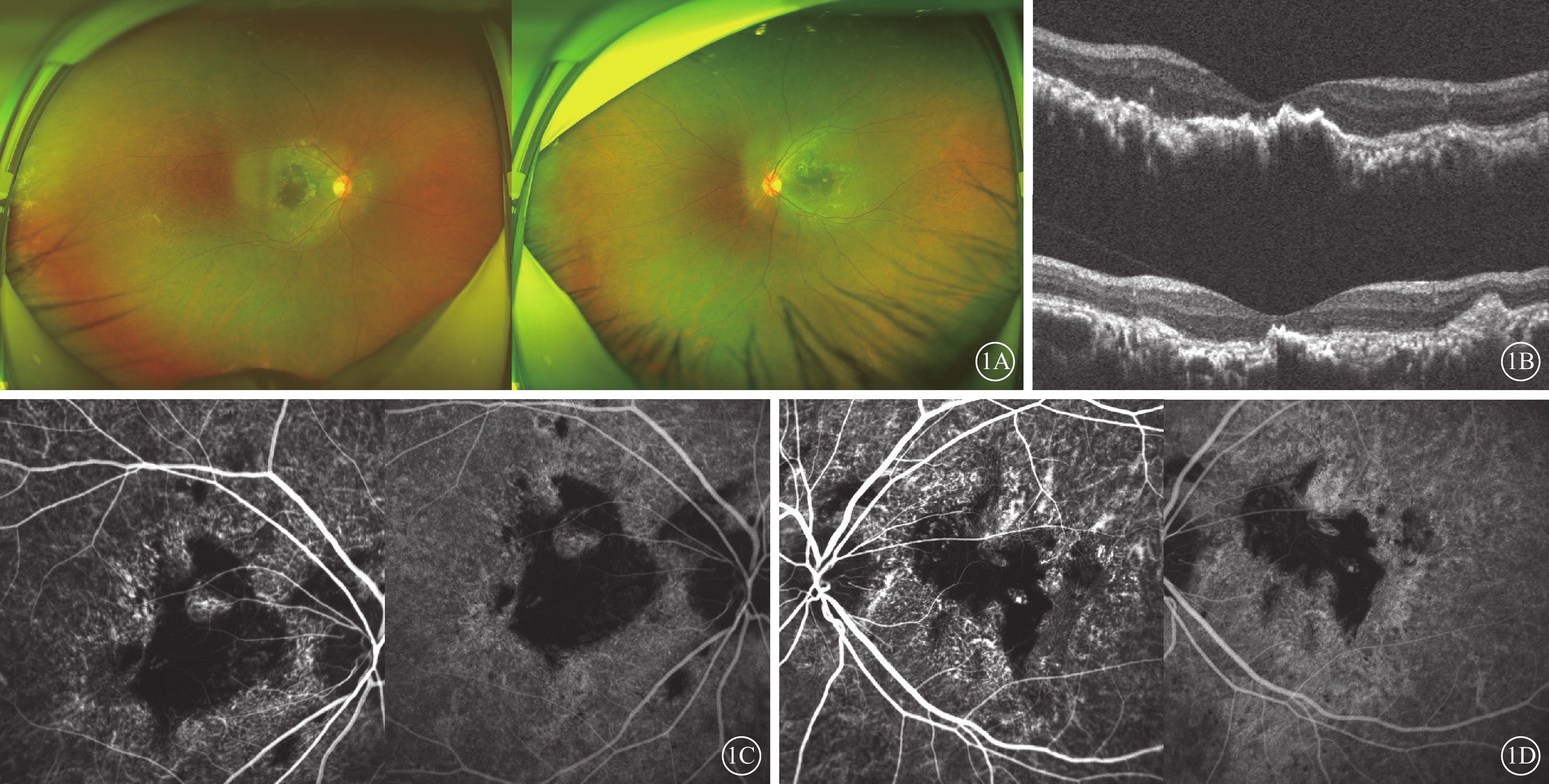
例1 患者女,36歲。因雙眼視力下降伴視物變形20余年,于2020年8月就診于昆明醫科大學第一附屬醫院眼科門診。父母非近親結婚,母親有“眼底出血”病史,外祖父和姐姐視力差。否認眼部外傷史、手術史等。眼部檢查:右眼裸眼視力0.01,矯正不能提高;左眼裸眼視力0.1,最佳矯正視力(BCVA)0.3。雙眼眼壓均為17 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見異常。眼底檢查,雙眼視盤邊界清楚,顏色淡紅,視網膜后極部以及黃斑血管弓范圍內可見中間深灰、周邊黃色,大片不規則的瘢痕組織形成,周邊視網膜可見點狀色素顆粒沉著,后極部血管走形正常,未見出血等改變(圖1A,1B)。光相干斷層掃描(OCT)檢查,雙眼黃斑區內層結構尚可,外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜結構凹凸不平、萎縮破壞,視網膜下大量團塊狀強反射信號(圖1C,1D)。吲哚青綠血管造影(ICGA)檢查,雙眼后極部視網膜可見地圖樣不規則弱熒光(圖1E~1H)。全身檢查及免疫等實驗室輔助檢查未見異常。臨床診斷:雙眼黃斑地圖樣萎縮。
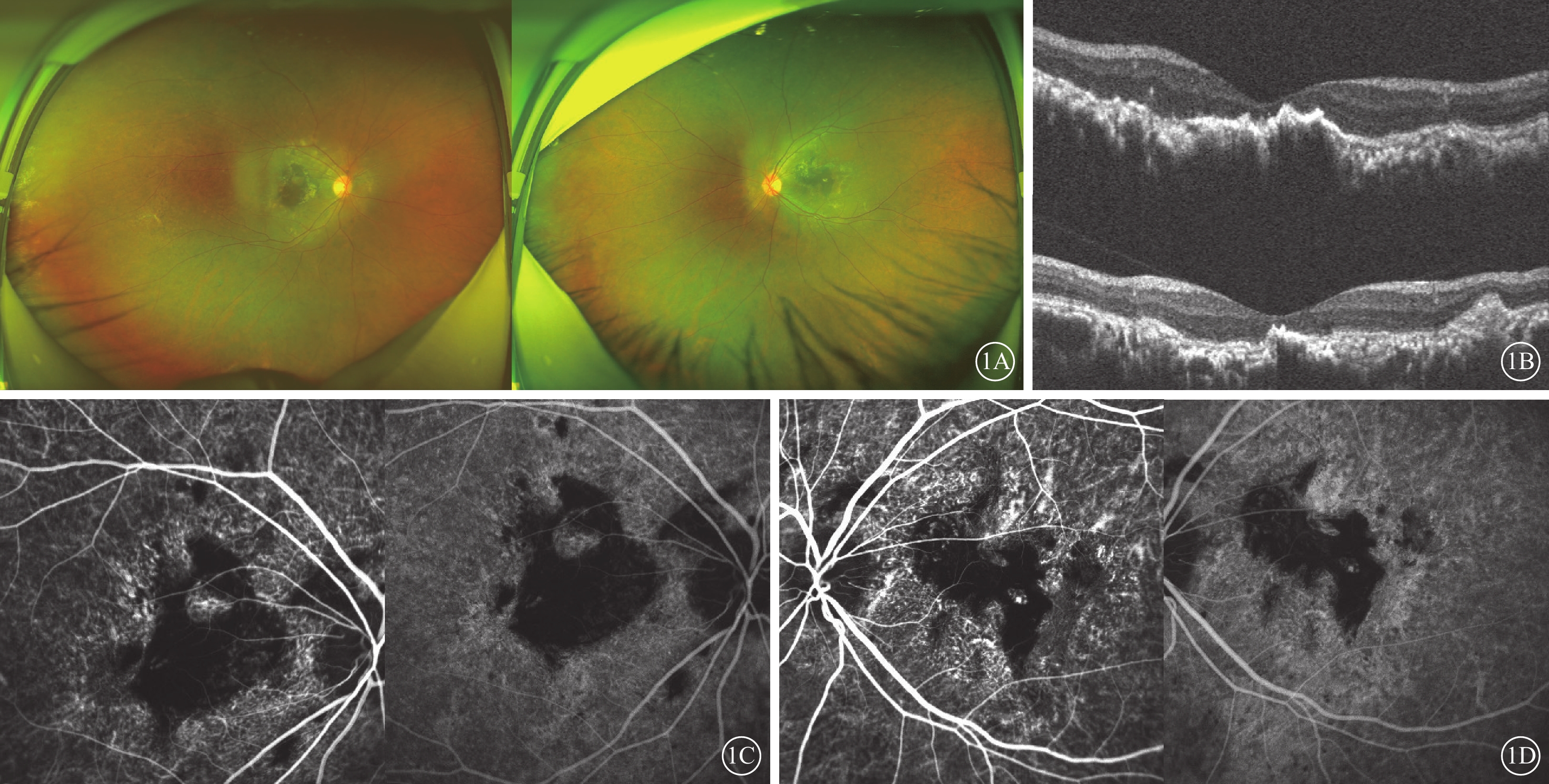 圖1
Sorsby眼底營養不良患者(例1患者)眼部檢查像 1A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。雙眼后極部黃斑區不規則斑片狀黃白色瘢痕灶,伴有色素增生、沉著。1B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區內層結構尚可,外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜結構凹凸不平、萎縮破壞,視網膜下大量團塊狀強反射信號。1C、1D分別示右眼、左眼吲哚青綠血管造影像,左圖為早期,右圖為晚期。雙眼黃斑區瘢痕性弱熒光及色素性遮蔽熒光,病灶內散在片狀強熒光,病灶周邊及后極部可見透見熒光,脈絡膜毛細血管萎縮
圖1
Sorsby眼底營養不良患者(例1患者)眼部檢查像 1A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。雙眼后極部黃斑區不規則斑片狀黃白色瘢痕灶,伴有色素增生、沉著。1B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區內層結構尚可,外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜結構凹凸不平、萎縮破壞,視網膜下大量團塊狀強反射信號。1C、1D分別示右眼、左眼吲哚青綠血管造影像,左圖為早期,右圖為晚期。雙眼黃斑區瘢痕性弱熒光及色素性遮蔽熒光,病灶內散在片狀強熒光,病灶周邊及后極部可見透見熒光,脈絡膜毛細血管萎縮
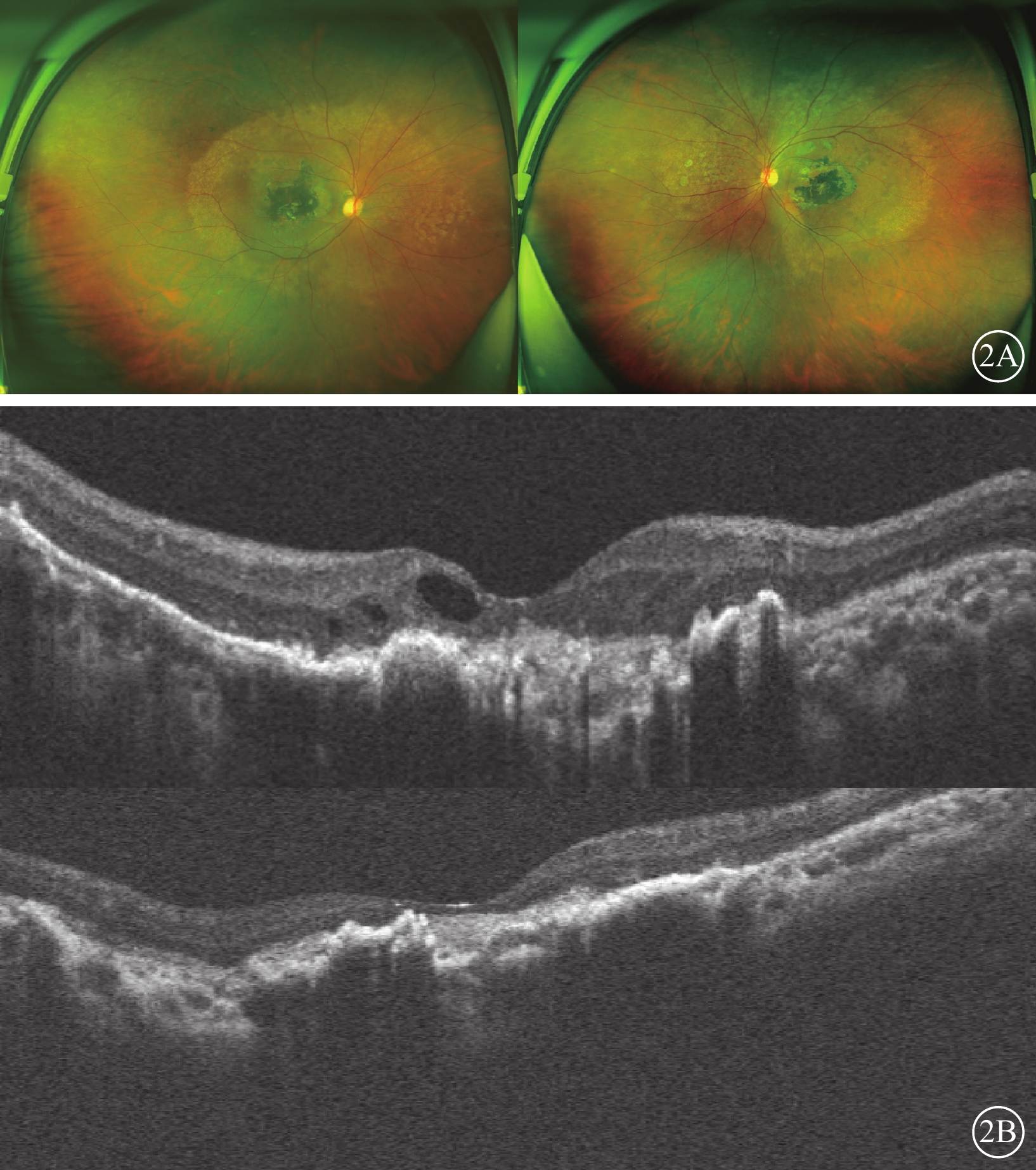
例2 患者女,40歲,例1患者姐姐。雙眼視力下降15年,于2020年8月就診于昆明醫科大學第一附屬醫院眼科門診。眼部檢查:右眼裸眼視力0.02,BCVA 0.05;左眼裸眼視力0.01,BCVA 0.04。右眼、左眼眼壓分別為17、18 mm Hg。雙眼眼前節未見異常。眼底檢查,黃斑區約4個視盤直徑(DD)大小的不規則斑片狀深灰色瘢痕、萎縮灶,伴大量色素增生、沉著;雙眼視盤邊界清楚,顏色淡紅,后極部血管走形正常,未見出血等改變。掃描激光檢眼鏡檢查,黃斑區4 DD大小疤痕樣萎縮灶,黃斑血管弓范圍可見點狀脫色素樣改變(圖2A)。OCT檢查,雙眼黃斑區外界膜、橢圓體帶、視網膜色素上皮層、Bruch膜、脈絡膜層結構破壞,右眼黃斑區萎縮呈強反射信號,視網膜神經上皮層間囊樣改變,左眼后極部視網膜萎縮變薄(圖2B)。全身檢查及免疫等實驗室輔助檢查未見異常。臨床診斷:雙眼黃斑地圖樣萎縮。
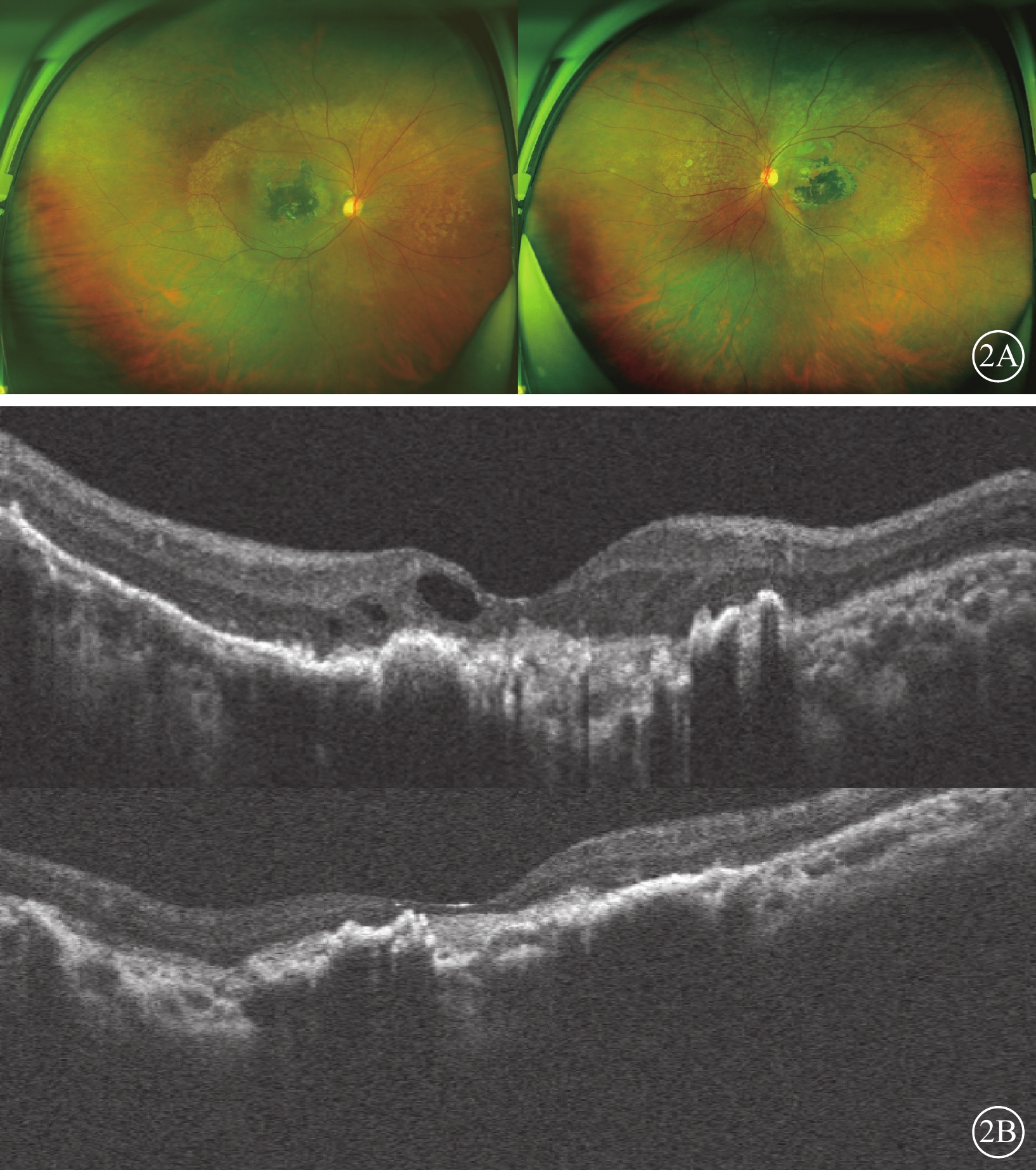 圖2
Sorsby眼底營養不良患者(例2患者,例1患者姐姐)眼部檢查像 2A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。視網膜色素分布不均勻,萎縮病灶大小約4個視盤直徑,集中于黃斑區,黃斑血管弓范圍可見點狀色素脫失。2B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜層結構破壞,右眼黃斑區萎縮呈強反射信號,視網膜神經上皮層間囊樣改變,左眼后極部視網膜萎縮變薄
圖2
Sorsby眼底營養不良患者(例2患者,例1患者姐姐)眼部檢查像 2A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。視網膜色素分布不均勻,萎縮病灶大小約4個視盤直徑,集中于黃斑區,黃斑血管弓范圍可見點狀色素脫失。2B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜層結構破壞,右眼黃斑區萎縮呈強反射信號,視網膜神經上皮層間囊樣改變,左眼后極部視網膜萎縮變薄
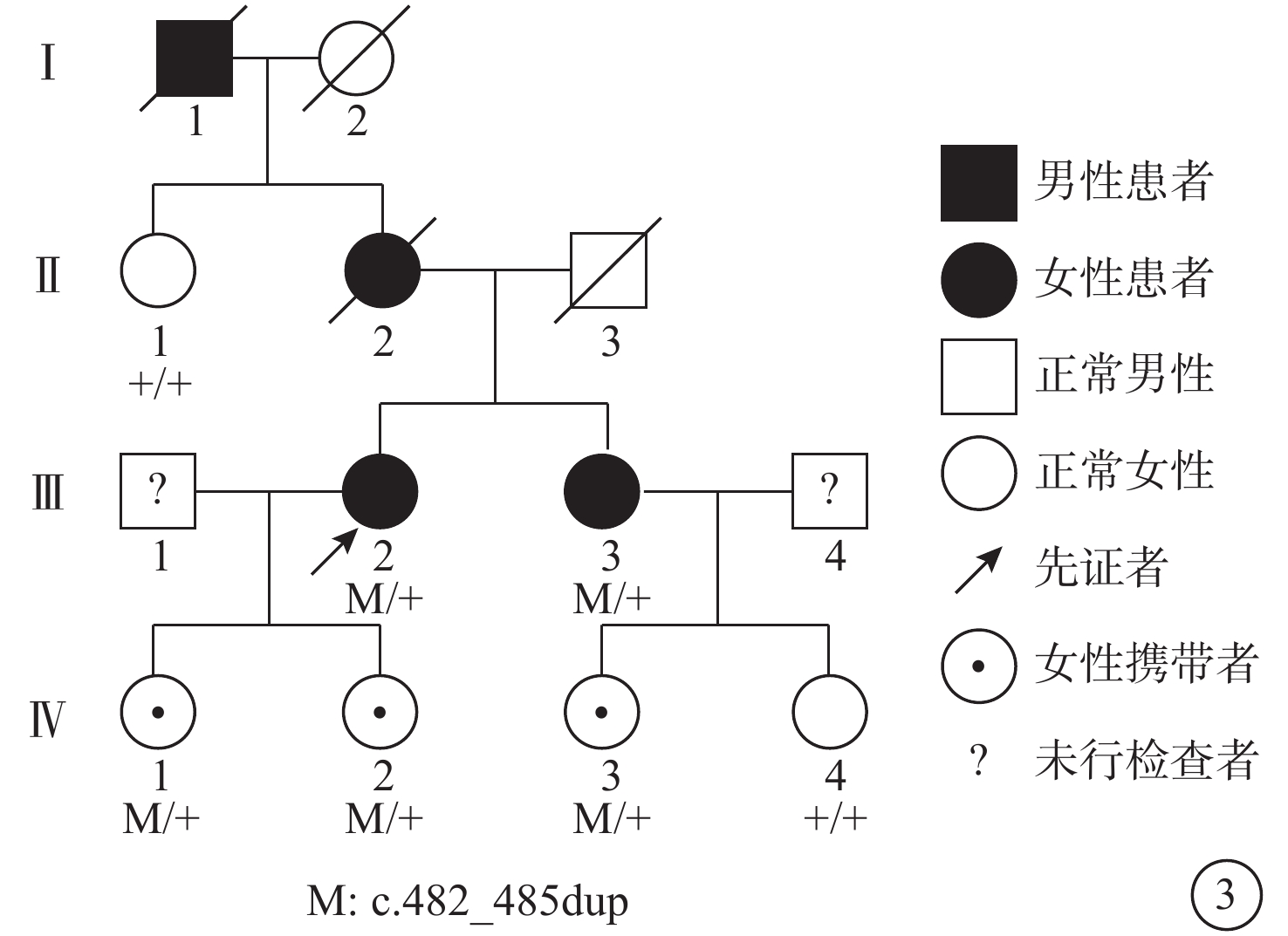
進一步追問病史,例1患者10年前曾因雙眼視力下降至當地醫院就診,考慮為“黃斑病變”,未予治療。由于本家系患者此次就診脈絡膜新生血管(CNV)已形成瘢痕,故對其采用口服葉黃素改善視覺質量,并進行定期隨訪,其他暫無特殊處理。對兩例患者的子女進行眼部檢查,暫未發現異常。結合患者姐姐類似眼底表現及母親與外祖父視力差的家族史,為明確診斷,采集兩例患者及其子女的全血樣本進行基因檢測。結果顯示,患者及兩女和患者姐姐及一女存在基因TIMP3(c.482-485dup)雜合突變,染色體位置chr22:32859223-32859226(圖3,4)。該變異為移碼變異重復變異,使得編碼蛋白在5號外顯子提前終止,影響了編碼蛋白超過10%的蛋白序列,很可能會影響蛋白功能。保守性分析顯示,TIMP3基因第5號外顯子編碼的第163位氨基酸位點在多物種間高度保守。該變異在人群基因頻率數據庫普通人數據庫東亞人群中頻率是0。根據美國醫學遺傳學與基因組學學會指南分析,突變具有1個PVS1證據、1個PM2證據、1個PP1證據、1個PP4證據,該突變可判為可能致病性突變。結合患者病史、臨床表現、影像學檢查和基因檢測結果,考慮診斷:Sorsby眼底營養不良(SFD)。
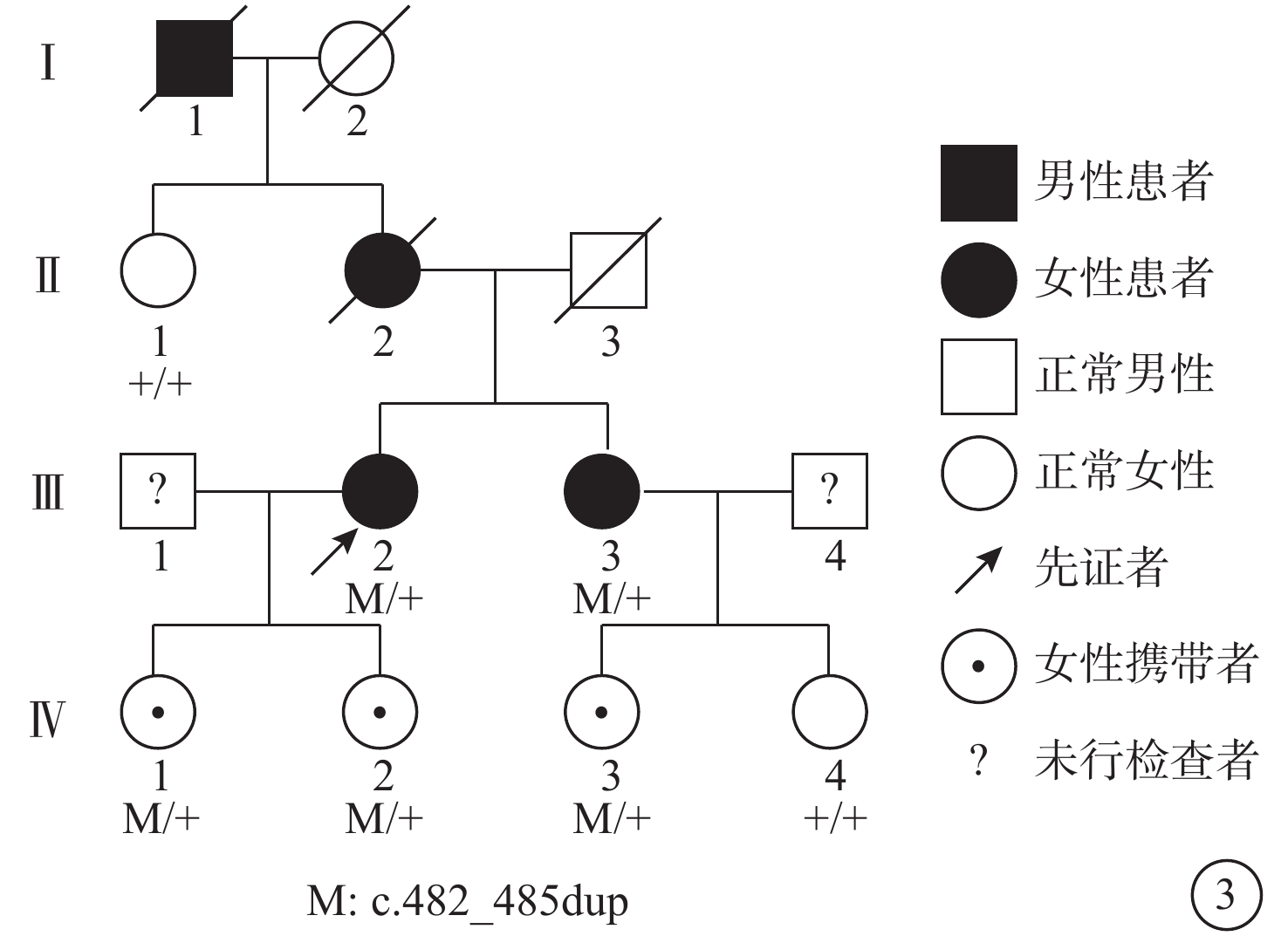 圖3
Sorsby眼底營養不良患者家系圖
圖3
Sorsby眼底營養不良患者家系圖
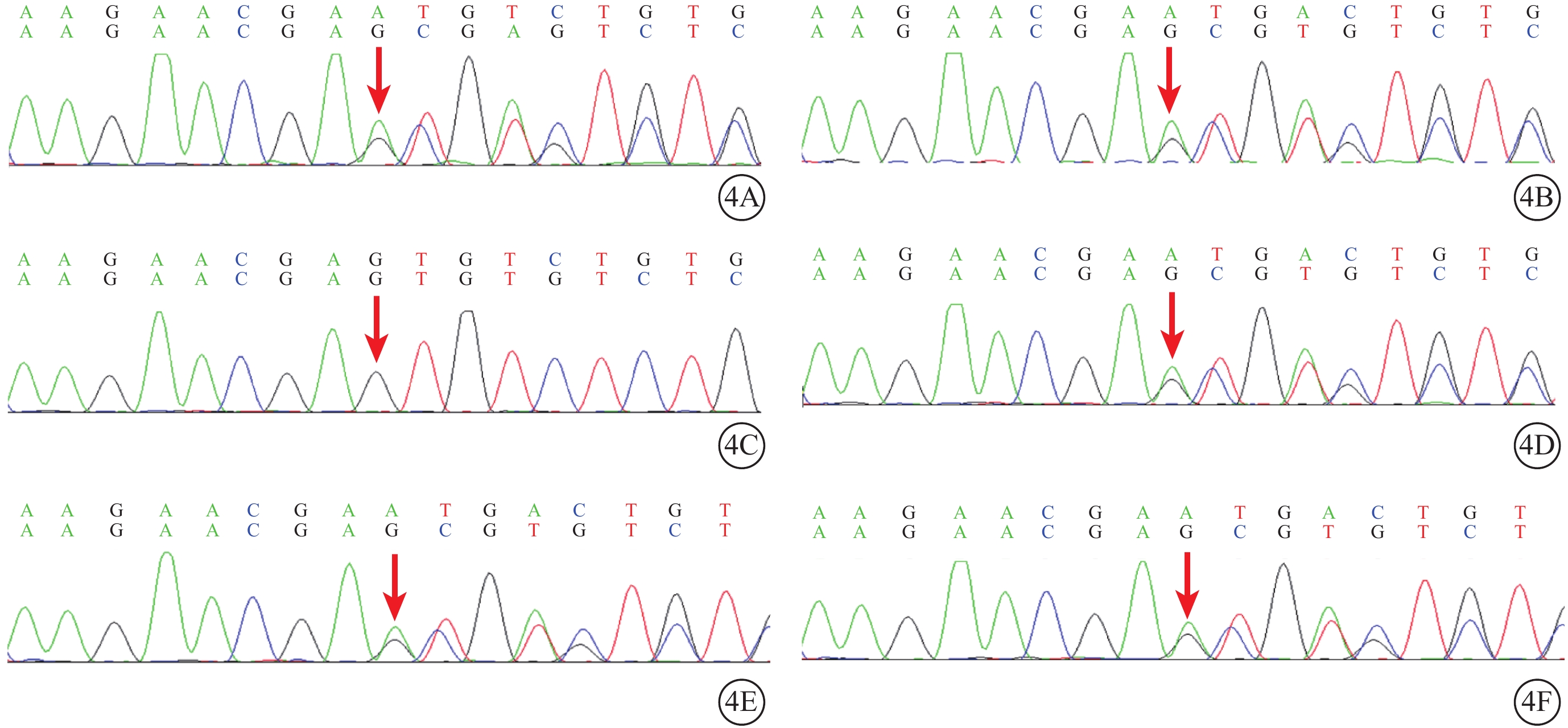 圖4
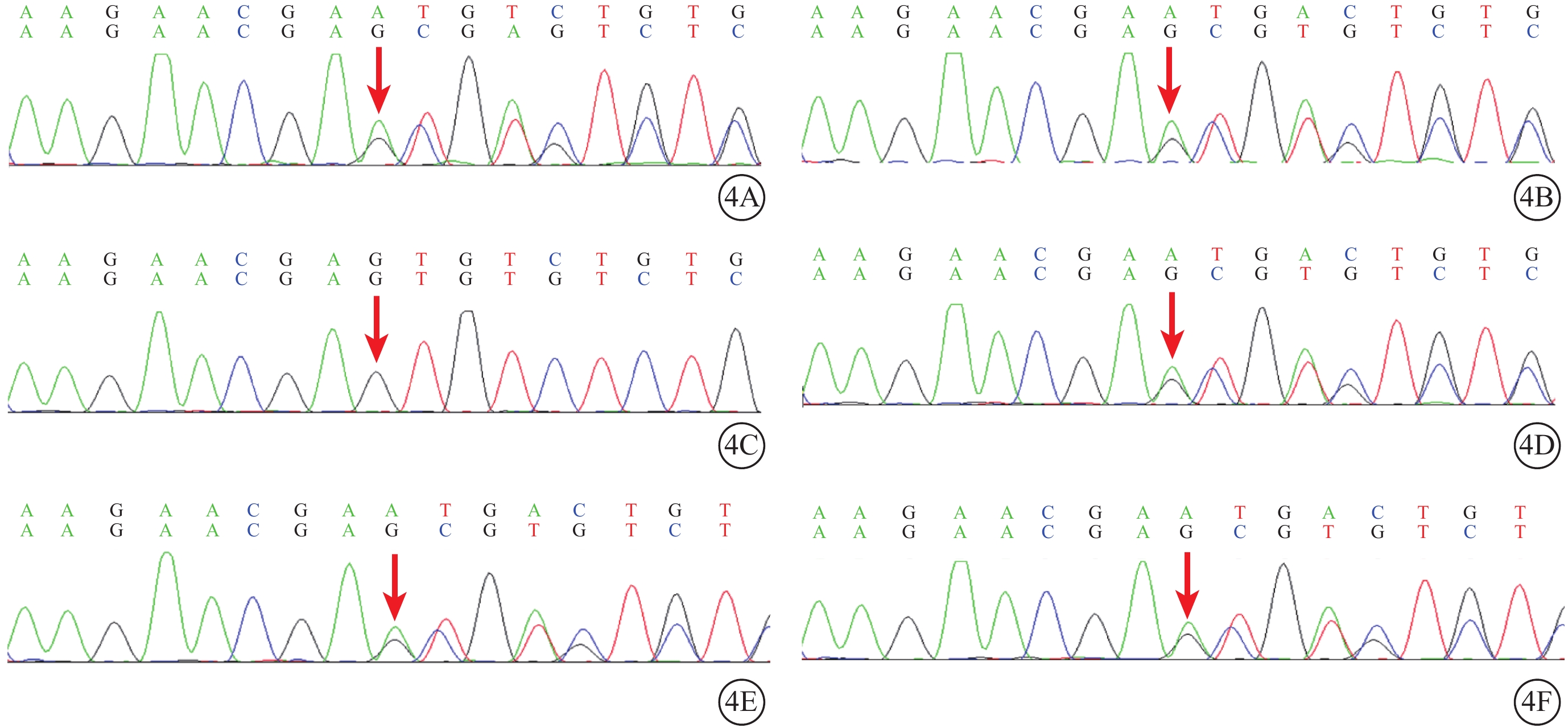
Sorsby眼底營養不良家系兩例患者及其子女基因測序圖 4A、4B分別示先證者(Ⅲ2)及其姐姐(Ⅲ3),均存在TIMP3基因第5號外顯子存在c.482-485dup:p.C163fs移碼變異(紅箭);4C示例2患者一女(Ⅳ4),該位點不存在突變;4D~4F分別示例2患者另一女(Ⅳ3)及例1患者兩女(Ⅳ1、Ⅳ2),均攜帶該位點突變(紅箭)
圖4
Sorsby眼底營養不良家系兩例患者及其子女基因測序圖 4A、4B分別示先證者(Ⅲ2)及其姐姐(Ⅲ3),均存在TIMP3基因第5號外顯子存在c.482-485dup:p.C163fs移碼變異(紅箭);4C示例2患者一女(Ⅳ4),該位點不存在突變;4D~4F分別示例2患者另一女(Ⅳ3)及例1患者兩女(Ⅳ1、Ⅳ2),均攜帶該位點突變(紅箭)
討論 SFD是一種常染色體顯性遺傳的黃斑退行性疾病,患者通常于30~40歲因CNV而表現為夜盲或突然喪失視力[1]。典型的臨床癥狀包括視物變形、中央暗點、色覺減退和中央視覺突然喪失[2]。中央視覺的突然改變通常是由于CNV的形成,而較緩慢的進行性視力喪失則是由于地圖樣萎縮區域的擴大造成[3]。SFD的臨床特征與老年性黃斑變性相似,包括玻璃膜疣、CNV形成和中央地圖樣萎縮,但其發病早、遺傳性強、晚期累及周圍脈絡膜視網膜造成萎縮是重要的鑒別特征。基因檢測通常是確診及臨床鑒別診斷的關鍵。本家系例1、例2患者可于后極部見到黃斑區不規則斑塊狀萎縮灶,周邊圍繞點狀色素沉積,視網膜色素上皮基底膜與Bruch膜之間的沉積物、CNV等表現,符合SFD的臨床表現特征[4]。結合本家系患者明確家族史及基因檢測結果,SFD診斷明確。
SFD的生理病理機制尚不明確,目前已知是TIMP3基因的特定突變引起[5-7]。TIMP3基因突變引起蛋白質結構的改變和聚集,可能導致了大量異常高分子蛋白質復合物在Bruch膜上積聚,導致運輸代謝物和營養物質的通透性降低,同時減弱了TIMP3對基質金屬蛋白酶和ADAM17的抑制作用,誘發血管的形成,產生CNV[8-9]。
本家系患者中,先證者(Ⅲ2)及其姐姐(Ⅲ3)均出現了雙眼對稱的黃斑區瘢痕樣萎縮灶,嚴重影響生活質量;而另外3例攜帶TIMP3基因突變的家系成員(Ⅳ1~3)眼底均未出現異常,這可能是因為其尚未到SFD 40~60歲的發病年齡[3]。
關于SFD的治療,目前主要采用抗血管內皮生長因子(VEGF)藥物抑制CNV。一項薈萃研究中認為,在SFD繼發CNV的患者中,當出現首發癥狀而延遲接受抗VEGF藥物治療可導致視網膜下瘢痕的形成,預后較差,早期行抗VEGF藥物治療對患者預后是有利的[10]。通過對該家系的成員進行基因檢測,可早期發現、早期診斷、早期干預,從而最大程度保留其視力。
例1 患者女,36歲。因雙眼視力下降伴視物變形20余年,于2020年8月就診于昆明醫科大學第一附屬醫院眼科門診。父母非近親結婚,母親有“眼底出血”病史,外祖父和姐姐視力差。否認眼部外傷史、手術史等。眼部檢查:右眼裸眼視力0.01,矯正不能提高;左眼裸眼視力0.1,最佳矯正視力(BCVA)0.3。雙眼眼壓均為17 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見異常。眼底檢查,雙眼視盤邊界清楚,顏色淡紅,視網膜后極部以及黃斑血管弓范圍內可見中間深灰、周邊黃色,大片不規則的瘢痕組織形成,周邊視網膜可見點狀色素顆粒沉著,后極部血管走形正常,未見出血等改變(圖1A,1B)。光相干斷層掃描(OCT)檢查,雙眼黃斑區內層結構尚可,外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜結構凹凸不平、萎縮破壞,視網膜下大量團塊狀強反射信號(圖1C,1D)。吲哚青綠血管造影(ICGA)檢查,雙眼后極部視網膜可見地圖樣不規則弱熒光(圖1E~1H)。全身檢查及免疫等實驗室輔助檢查未見異常。臨床診斷:雙眼黃斑地圖樣萎縮。
圖1
Sorsby眼底營養不良患者(例1患者)眼部檢查像 1A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。雙眼后極部黃斑區不規則斑片狀黃白色瘢痕灶,伴有色素增生、沉著。1B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區內層結構尚可,外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜結構凹凸不平、萎縮破壞,視網膜下大量團塊狀強反射信號。1C、1D分別示右眼、左眼吲哚青綠血管造影像,左圖為早期,右圖為晚期。雙眼黃斑區瘢痕性弱熒光及色素性遮蔽熒光,病灶內散在片狀強熒光,病灶周邊及后極部可見透見熒光,脈絡膜毛細血管萎縮
例2 患者女,40歲,例1患者姐姐。雙眼視力下降15年,于2020年8月就診于昆明醫科大學第一附屬醫院眼科門診。眼部檢查:右眼裸眼視力0.02,BCVA 0.05;左眼裸眼視力0.01,BCVA 0.04。右眼、左眼眼壓分別為17、18 mm Hg。雙眼眼前節未見異常。眼底檢查,黃斑區約4個視盤直徑(DD)大小的不規則斑片狀深灰色瘢痕、萎縮灶,伴大量色素增生、沉著;雙眼視盤邊界清楚,顏色淡紅,后極部血管走形正常,未見出血等改變。掃描激光檢眼鏡檢查,黃斑區4 DD大小疤痕樣萎縮灶,黃斑血管弓范圍可見點狀脫色素樣改變(圖2A)。OCT檢查,雙眼黃斑區外界膜、橢圓體帶、視網膜色素上皮層、Bruch膜、脈絡膜層結構破壞,右眼黃斑區萎縮呈強反射信號,視網膜神經上皮層間囊樣改變,左眼后極部視網膜萎縮變薄(圖2B)。全身檢查及免疫等實驗室輔助檢查未見異常。臨床診斷:雙眼黃斑地圖樣萎縮。
圖2
Sorsby眼底營養不良患者(例2患者,例1患者姐姐)眼部檢查像 2A示雙眼廣角彩色眼底像,左圖為右眼,右圖為左眼。視網膜色素分布不均勻,萎縮病灶大小約4個視盤直徑,集中于黃斑區,黃斑血管弓范圍可見點狀色素脫失。2B示光相干斷層掃描像,上圖為右眼,下圖為左眼。雙眼黃斑區外界膜、橢圓體帶、視網膜色素上皮、Bruch膜、脈絡膜層結構破壞,右眼黃斑區萎縮呈強反射信號,視網膜神經上皮層間囊樣改變,左眼后極部視網膜萎縮變薄
進一步追問病史,例1患者10年前曾因雙眼視力下降至當地醫院就診,考慮為“黃斑病變”,未予治療。由于本家系患者此次就診脈絡膜新生血管(CNV)已形成瘢痕,故對其采用口服葉黃素改善視覺質量,并進行定期隨訪,其他暫無特殊處理。對兩例患者的子女進行眼部檢查,暫未發現異常。結合患者姐姐類似眼底表現及母親與外祖父視力差的家族史,為明確診斷,采集兩例患者及其子女的全血樣本進行基因檢測。結果顯示,患者及兩女和患者姐姐及一女存在基因TIMP3(c.482-485dup)雜合突變,染色體位置chr22:32859223-32859226(圖3,4)。該變異為移碼變異重復變異,使得編碼蛋白在5號外顯子提前終止,影響了編碼蛋白超過10%的蛋白序列,很可能會影響蛋白功能。保守性分析顯示,TIMP3基因第5號外顯子編碼的第163位氨基酸位點在多物種間高度保守。該變異在人群基因頻率數據庫普通人數據庫東亞人群中頻率是0。根據美國醫學遺傳學與基因組學學會指南分析,突變具有1個PVS1證據、1個PM2證據、1個PP1證據、1個PP4證據,該突變可判為可能致病性突變。結合患者病史、臨床表現、影像學檢查和基因檢測結果,考慮診斷:Sorsby眼底營養不良(SFD)。
圖3
Sorsby眼底營養不良患者家系圖
圖4
Sorsby眼底營養不良家系兩例患者及其子女基因測序圖 4A、4B分別示先證者(Ⅲ2)及其姐姐(Ⅲ3),均存在TIMP3基因第5號外顯子存在c.482-485dup:p.C163fs移碼變異(紅箭);4C示例2患者一女(Ⅳ4),該位點不存在突變;4D~4F分別示例2患者另一女(Ⅳ3)及例1患者兩女(Ⅳ1、Ⅳ2),均攜帶該位點突變(紅箭)
討論 SFD是一種常染色體顯性遺傳的黃斑退行性疾病,患者通常于30~40歲因CNV而表現為夜盲或突然喪失視力[1]。典型的臨床癥狀包括視物變形、中央暗點、色覺減退和中央視覺突然喪失[2]。中央視覺的突然改變通常是由于CNV的形成,而較緩慢的進行性視力喪失則是由于地圖樣萎縮區域的擴大造成[3]。SFD的臨床特征與老年性黃斑變性相似,包括玻璃膜疣、CNV形成和中央地圖樣萎縮,但其發病早、遺傳性強、晚期累及周圍脈絡膜視網膜造成萎縮是重要的鑒別特征。基因檢測通常是確診及臨床鑒別診斷的關鍵。本家系例1、例2患者可于后極部見到黃斑區不規則斑塊狀萎縮灶,周邊圍繞點狀色素沉積,視網膜色素上皮基底膜與Bruch膜之間的沉積物、CNV等表現,符合SFD的臨床表現特征[4]。結合本家系患者明確家族史及基因檢測結果,SFD診斷明確。
SFD的生理病理機制尚不明確,目前已知是TIMP3基因的特定突變引起[5-7]。TIMP3基因突變引起蛋白質結構的改變和聚集,可能導致了大量異常高分子蛋白質復合物在Bruch膜上積聚,導致運輸代謝物和營養物質的通透性降低,同時減弱了TIMP3對基質金屬蛋白酶和ADAM17的抑制作用,誘發血管的形成,產生CNV[8-9]。
本家系患者中,先證者(Ⅲ2)及其姐姐(Ⅲ3)均出現了雙眼對稱的黃斑區瘢痕樣萎縮灶,嚴重影響生活質量;而另外3例攜帶TIMP3基因突變的家系成員(Ⅳ1~3)眼底均未出現異常,這可能是因為其尚未到SFD 40~60歲的發病年齡[3]。
關于SFD的治療,目前主要采用抗血管內皮生長因子(VEGF)藥物抑制CNV。一項薈萃研究中認為,在SFD繼發CNV的患者中,當出現首發癥狀而延遲接受抗VEGF藥物治療可導致視網膜下瘢痕的形成,預后較差,早期行抗VEGF藥物治療對患者預后是有利的[10]。通過對該家系的成員進行基因檢測,可早期發現、早期診斷、早期干預,從而最大程度保留其視力。