引用本文: 李瑞梅, 周慧, 李雙農. Coats樣視網膜色素變性1例. 中華眼底病雜志, 2023, 39(7): 591-593. doi: 10.3760/cma.j.cn511434-20220411-00210 復制
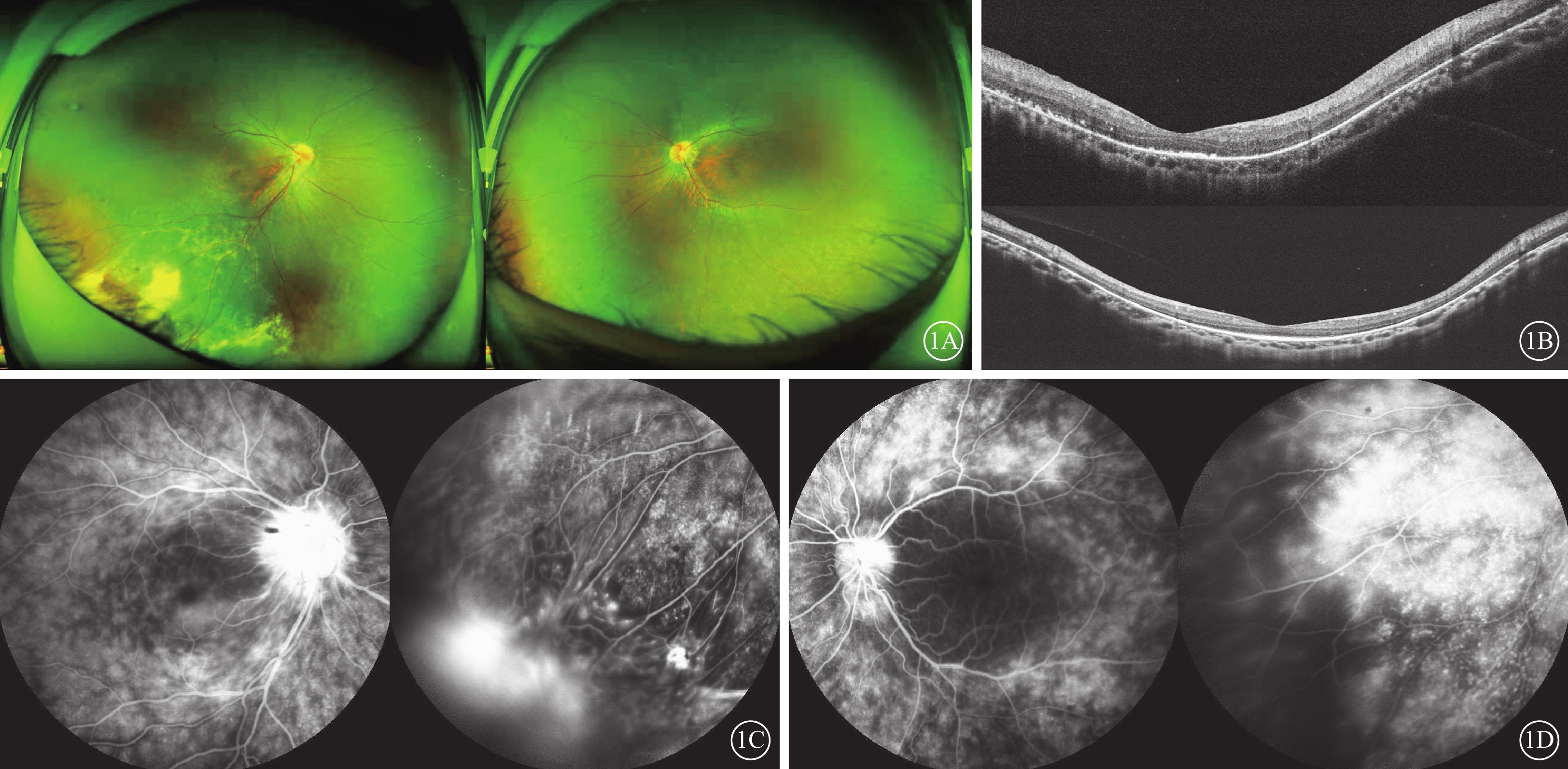
患者男,16歲。因右眼視物模糊半年于2020年10月9日在山西愛爾眼科醫院就診。半年前于外院診斷為“右眼Coats病”并行視網膜激光光凝治療。否認既往眼部病史及全身特殊病史,否認家族遺傳病史。眼部檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.3、0.5。右眼、左眼眼壓均為16 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見明顯異常,玻璃體混濁(+),細胞(+)。眼底檢查:雙眼視盤邊界欠清,顏色略紅,視網膜血管較細,周邊視網膜色素分布不均,無色素游離;右眼顳下方視網膜下有黃白色脂質滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血(圖1A)。光相干斷層掃描檢查,右眼以外層結構破壞為主,視網膜色素上皮(RPE)表面可見顆粒樣強反射,除中心凹處橢圓體帶不連續外,其余部位橢圓體帶消失;左眼中心凹處橢圓體帶結構不清晰,其余部位橢圓體帶消失(圖1B)。熒光素眼底血管造影(FFA)檢查,雙眼視盤強熒光,深層毛細血管及周邊視網膜血管熒光素滲漏,右眼顳下方血管閉塞,見小片狀無灌注(圖1C,1D)。結核、風濕免疫相關檢查結果均為陰性。臨床初步診斷:雙眼視網膜血管炎。
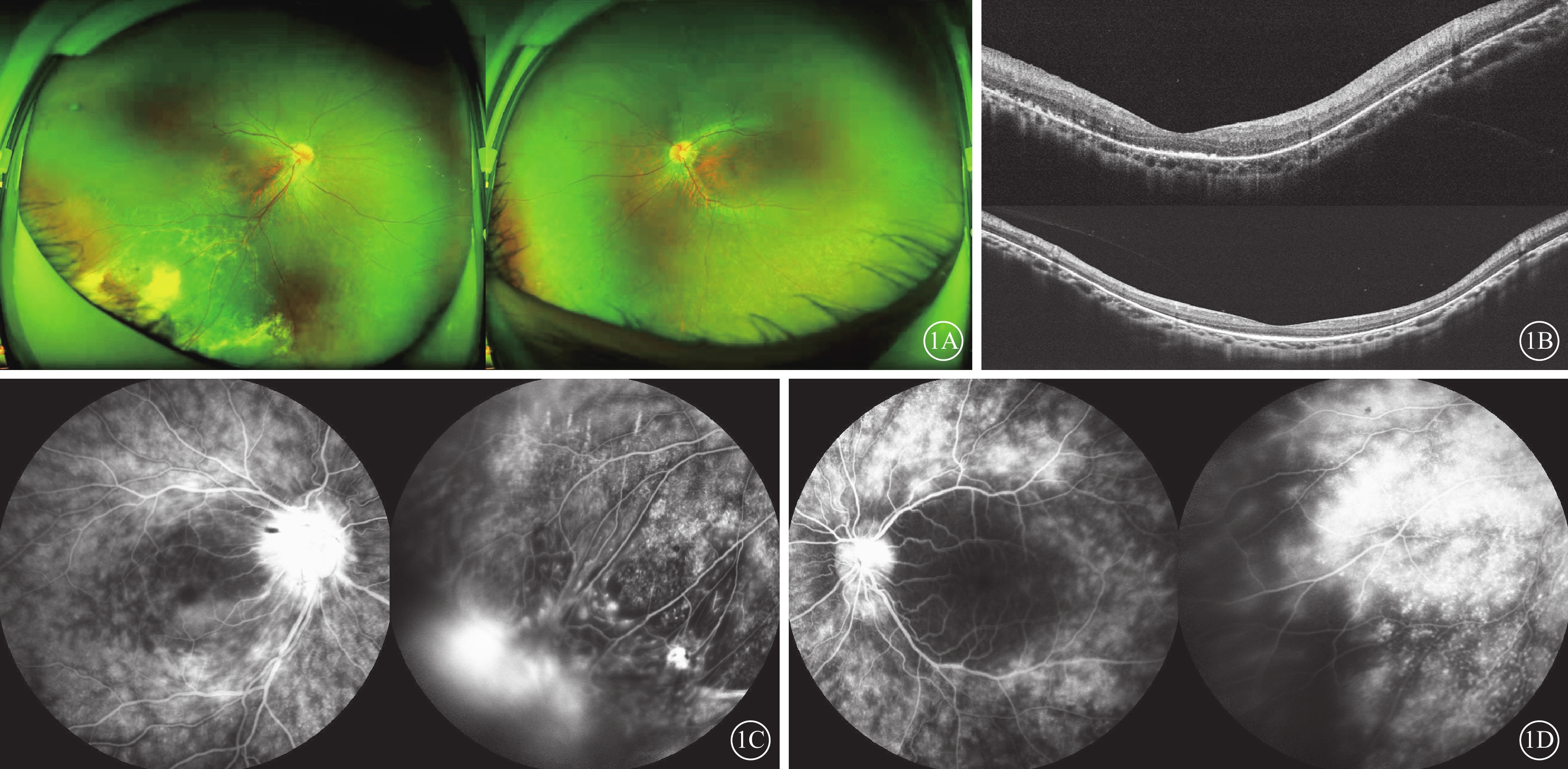 圖1
Coats樣視網膜色素變性患者初診時眼部檢查像 1A示雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視盤邊界欠清,顏色略紅,視網膜血管較細,周邊視網膜色素分布不均,呈椒鹽樣外觀,無色素游離;右眼顳下方視網膜下有黃白色脂性滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血。1B示雙眼光相干斷層掃描像,上圖為右眼,下圖為左眼。右眼以外層結構破壞為主,視網膜色素上皮表面可見顆粒樣強反射,除中心凹處橢圓體帶不連續外,其余部位橢圓體帶消失;左眼中心凹處橢圓體帶結構不清晰,其余部位橢圓體帶消失。1C、1D分別示右眼、左眼熒光素眼底血管造影像。雙眼視盤呈強熒光,深層毛細血管滲漏,周邊視網膜血管滲漏;右眼顳下方血管閉塞,見小片狀無灌注
圖1
Coats樣視網膜色素變性患者初診時眼部檢查像 1A示雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視盤邊界欠清,顏色略紅,視網膜血管較細,周邊視網膜色素分布不均,呈椒鹽樣外觀,無色素游離;右眼顳下方視網膜下有黃白色脂性滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血。1B示雙眼光相干斷層掃描像,上圖為右眼,下圖為左眼。右眼以外層結構破壞為主,視網膜色素上皮表面可見顆粒樣強反射,除中心凹處橢圓體帶不連續外,其余部位橢圓體帶消失;左眼中心凹處橢圓體帶結構不清晰,其余部位橢圓體帶消失。1C、1D分別示右眼、左眼熒光素眼底血管造影像。雙眼視盤呈強熒光,深層毛細血管滲漏,周邊視網膜血管滲漏;右眼顳下方血管閉塞,見小片狀無灌注
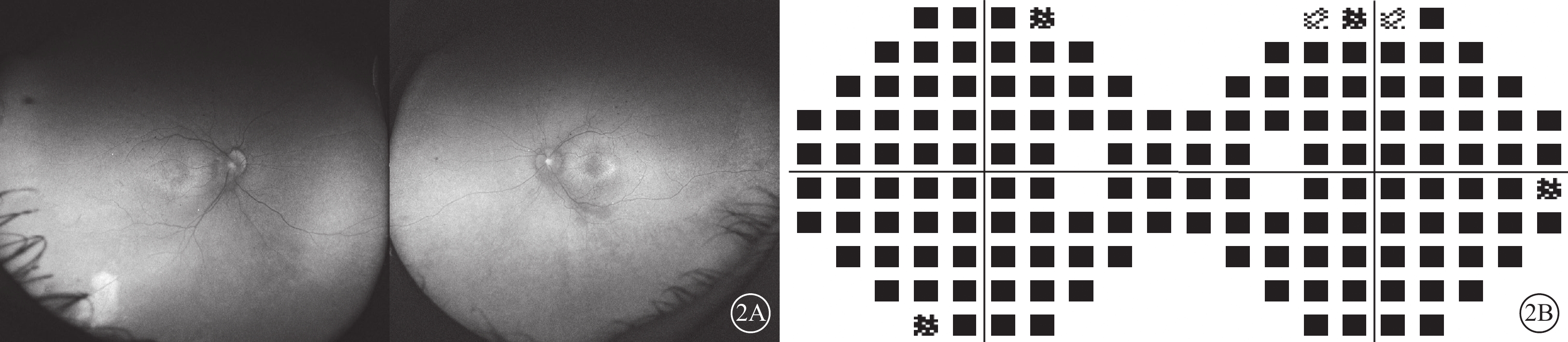
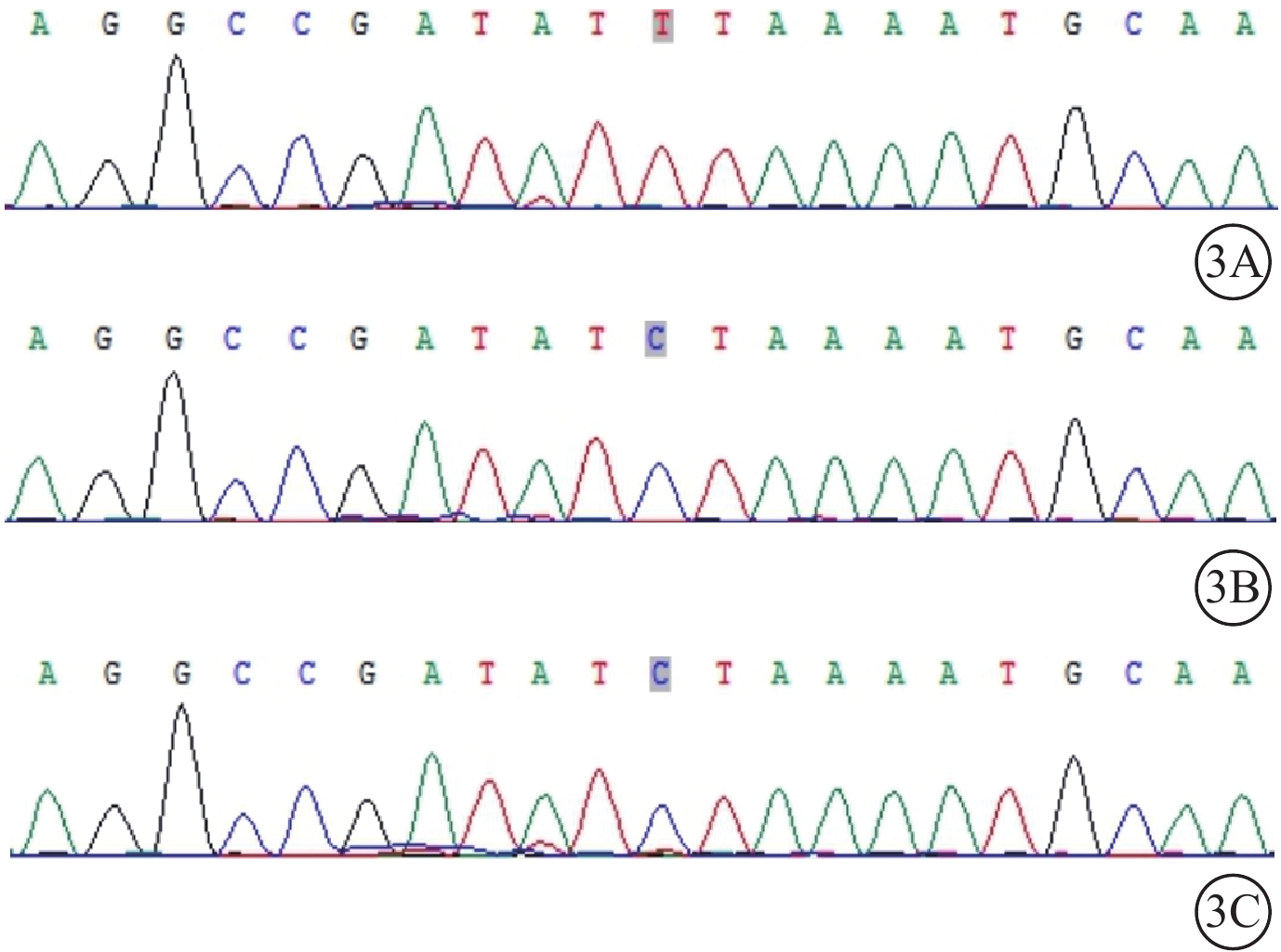
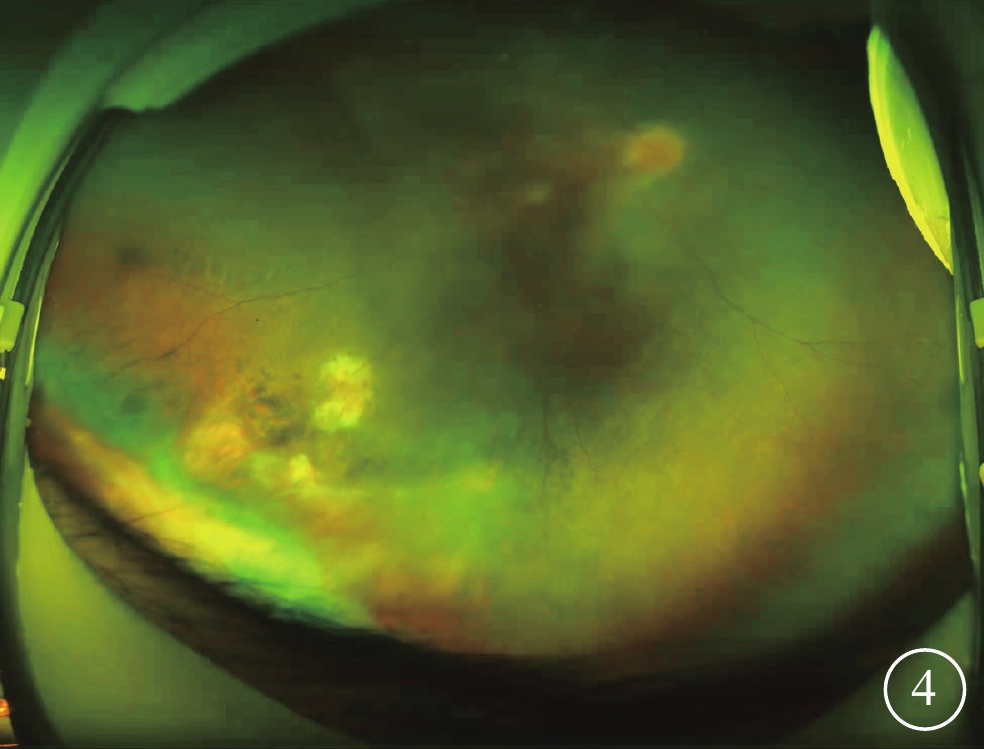
給予患者右眼后Tenon囊下注射曲安奈德20 mg。患者失訪4個月后復診:視力穩定,眼部檢查無明顯變化。考慮患者病情轉歸與視網膜血管炎的特征不相符,追問病史,患者訴自幼夜盲史。進一步完善相關檢查。眼底自身熒光檢查,雙眼中周部熒光變暗(圖2A)。視野檢查,雙眼明顯向心性縮窄(圖2B)。視網膜電圖檢查,雙眼明、暗視反應均未引出波形。結合患者眼底表現,考慮視網膜色素變性(RP)可能。對患者及其父母外周血行基因檢測(北京中因科技有限公司),結果顯示患者攜帶半合子變異,系RPGR基因c.935-1G>A位點突變(圖3)。其父親、母親均為野生型。最終診斷:雙眼X性連鎖RP(XLRP)合并右眼Coats樣改變。給予患者右眼顳下方視網膜異常血管病灶處行鞏膜外冷凍聯合后Tenon囊下注射曲安奈德20 mg治療,2周后對顳下方補充視網膜激光光凝。治療后4個月復診,右眼、左眼BCVA分別為0.3、0.5;患者右眼顳下方視網膜病灶區血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小(圖4)。隨訪期間曾出現一過性高眼壓,藥物能夠控制。
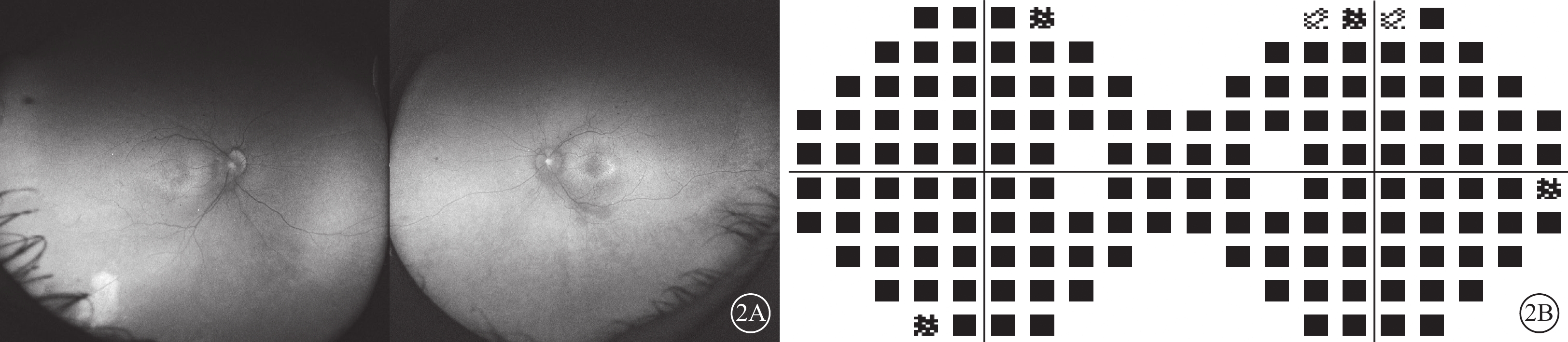 圖2
Coats樣視網膜色素變性患者復診時眼部檢查像 2A示眼底自身熒光像,左圖為右眼,右圖為左眼。雙眼中周部熒光變暗。2B示視野像,左圖為右眼,右圖為左眼。雙眼視野向心性狹窄
圖2
Coats樣視網膜色素變性患者復診時眼部檢查像 2A示眼底自身熒光像,左圖為右眼,右圖為左眼。雙眼中周部熒光變暗。2B示視野像,左圖為右眼,右圖為左眼。雙眼視野向心性狹窄
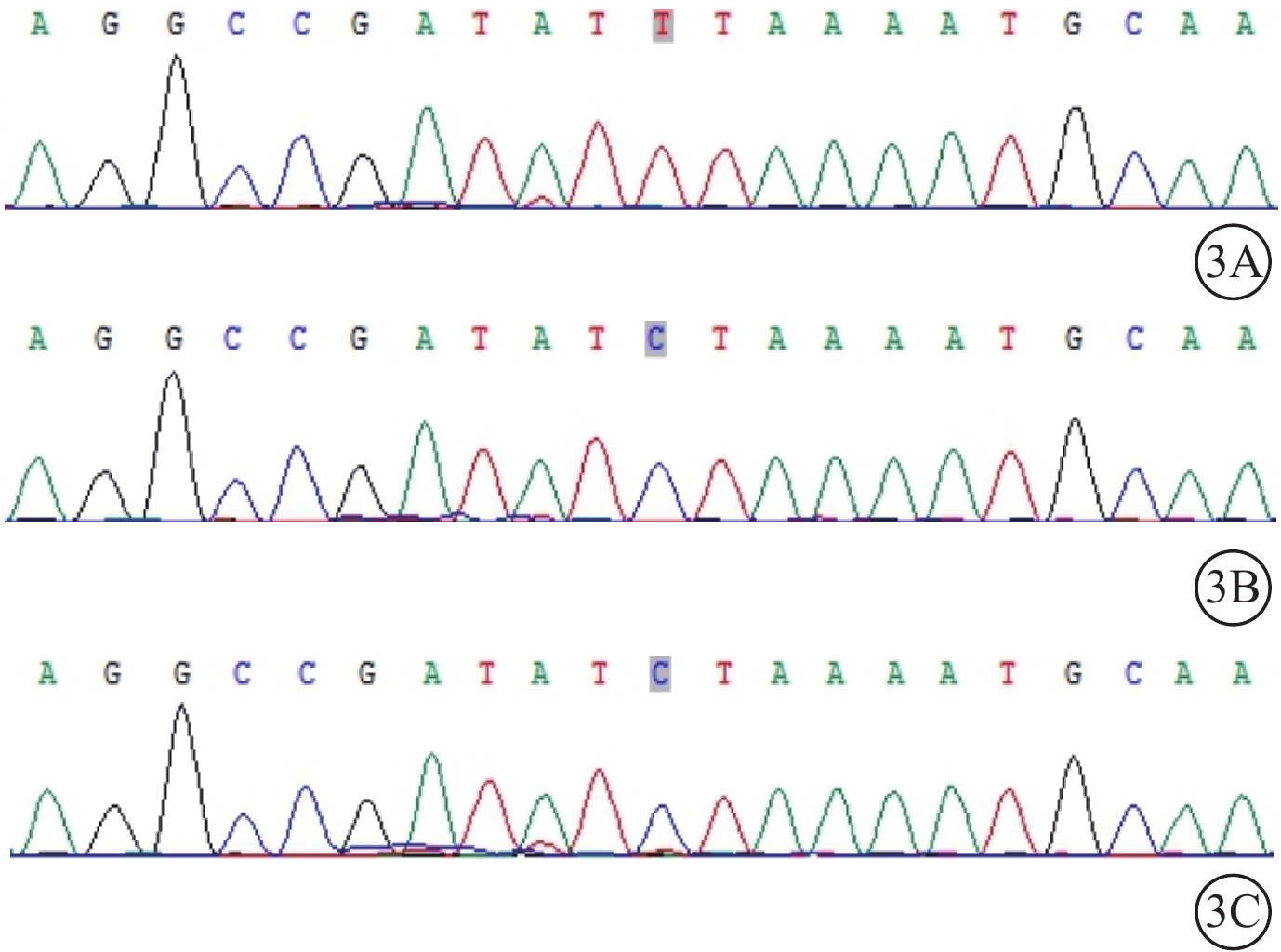 圖3
Coats樣視網膜色素變性患者及其父母基因測序圖 3A示患者,攜帶半合子變異,系RPGR基因c.935-1G>A位點突變;3B示患者父親;3C示患者母親。其父親、母親均為野生型
圖3
Coats樣視網膜色素變性患者及其父母基因測序圖 3A示患者,攜帶半合子變異,系RPGR基因c.935-1G>A位點突變;3B示患者父親;3C示患者母親。其父親、母親均為野生型
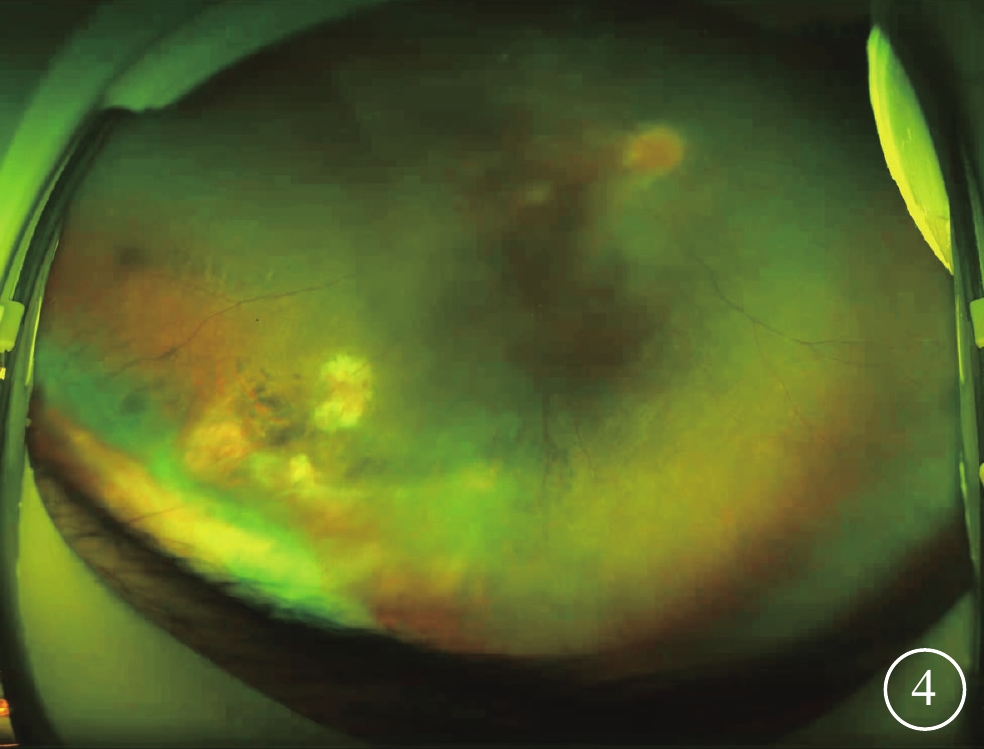 圖4
Coats樣視網膜色素變性患者治療后右眼彩色眼底像 顳下方視網膜病灶區血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小
圖4
Coats樣視網膜色素變性患者治療后右眼彩色眼底像 顳下方視網膜病灶區血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小
討論 RP是一組以光感受器細胞及RPE細胞進行性損害為特征的遺傳性疾病[1]。典型的RP眼底常常表現為視盤蠟黃、視網膜血管一致性狹窄、骨細胞樣色素沉著等,結合病史和其他檢查,通常不容易誤診。但是對于一些不典型的RP,如眼底缺乏骨細胞樣色素沉著,可伴有視網膜血管滲漏、甚至黃斑水腫者[2],可能導致誤診。本例患者玻璃體細胞(+),雙眼視盤顏色略紅,視網膜血管較細,周邊視網膜無明顯的色素游離;FFA檢查示雙眼視盤強熒光,深層毛細血管及周邊視網膜血管熒光素滲漏;有炎癥性表現,未關注到特征性RP的表現,因此首診時誤診為雙眼視網膜血管炎。
Coats樣RP目前文獻報道極少,其在RP中的發病率約1%~5%[3]。該病無明顯性別差異,常雙眼發病,呈對稱性;Coats樣改變通常在RP診斷以后,表現為富含脂性的漿液性視網膜脫離,表面血管扭曲擴張,病灶常位于赤道部或之前,局限于顳下象限,偶可累及后極部黃斑與視盤,且常為多灶性,滲出病變此消彼長;FFA檢查可見RPE點狀透見熒光,視網膜脫離處毛細血管擴張,微動脈瘤,視網膜新生血管形成,晚期熒光素滲漏,周邊無灌注,伴毛細血管擴張滲漏[3]。Coats樣RP常常需要與Coats病鑒別,尤其是眼底表現不典型的RP。Coats病是一種非遺傳性滲出性視網膜血管疾病,好發于年輕男性,單眼發病,主要表現為視網膜毛細血管擴張癥,微動脈瘤,視網膜內、下脂質沉積和纖維化,以及不同程度的滲出性視網膜脫離。根據患者的臨床表現、性別和發病特點,結合遺傳學特征,往往不難鑒別。本例患者自幼夜盲史;雙眼發病,視網膜血管較細,周邊視網膜色素分布不均;視野檢查示雙眼明顯向心性縮窄;ERG檢查示雙眼明、暗視反應均未引出波形;且基因檢測發現RPGR基因c.935-1G>A位點突變。即RP診斷成立。同時,本例患者右眼顳下方視網膜下有黃白色脂質滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血;再結合其FFA檢查表現,符合RP合并右眼Coats樣改變的診斷。
該病的發病機制可能是由于光感受器和RPE降解產生的有毒物質累積,誘發血管舒張反應,以及視網膜血管狹窄導致視網膜組織慢性缺血、缺氧,從而引發毛細血管擴張、微動脈瘤及視網膜新生血管形成[4-5]。
Coats樣RP的變異基因包括CRB1、RPGR、CRX、CEP290、PRPF8、CNGB1、TULP1等[3, 5],其中以CRB1突變報道較多,被認為是Coats樣RP的重要危險因素。Demirci等[6]報道了1例XLRP合并Coats樣病例,發現RPGR基因外顯子ORF15發生突變(912G>T)。本例患者致病基因為RPGR,c.935-1G>A 位點變異,其父親、母親均為野生型。該位點為新發突變位點,僅在ClinVar數據庫中收錄為致病性,在外顯子組聚集聯盟數據庫、人群基因頻率數據庫、人類基因致病性突變數據庫數據庫均未收錄。
目前Coats樣RP的治療主要針對視網膜異常血管病變導致的并發癥,輕者通常采用視網膜激光光凝、鞏膜外冷凍封閉異常血管病變;玻璃體腔注射抗血管內皮生長因子藥物減輕視網膜出血、滲出;對于伴有嚴重的視網膜脫離可行視網膜下液引流、鞏膜外墊壓,甚至玻璃體切割手術[3]。本例患者經鞏膜外冷凍封閉異常血管病變及后Tenon囊下注射曲安奈德治療,同時后期輔以視網膜激光光凝補充治療后,視網膜血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小。
患者男,16歲。因右眼視物模糊半年于2020年10月9日在山西愛爾眼科醫院就診。半年前于外院診斷為“右眼Coats病”并行視網膜激光光凝治療。否認既往眼部病史及全身特殊病史,否認家族遺傳病史。眼部檢查:右眼、左眼最佳矯正視力(BCVA)分別為0.3、0.5。右眼、左眼眼壓均為16 mm Hg(1 mm Hg=0.133 kPa)。雙眼眼前節未見明顯異常,玻璃體混濁(+),細胞(+)。眼底檢查:雙眼視盤邊界欠清,顏色略紅,視網膜血管較細,周邊視網膜色素分布不均,無色素游離;右眼顳下方視網膜下有黃白色脂質滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血(圖1A)。光相干斷層掃描檢查,右眼以外層結構破壞為主,視網膜色素上皮(RPE)表面可見顆粒樣強反射,除中心凹處橢圓體帶不連續外,其余部位橢圓體帶消失;左眼中心凹處橢圓體帶結構不清晰,其余部位橢圓體帶消失(圖1B)。熒光素眼底血管造影(FFA)檢查,雙眼視盤強熒光,深層毛細血管及周邊視網膜血管熒光素滲漏,右眼顳下方血管閉塞,見小片狀無灌注(圖1C,1D)。結核、風濕免疫相關檢查結果均為陰性。臨床初步診斷:雙眼視網膜血管炎。
圖1
Coats樣視網膜色素變性患者初診時眼部檢查像 1A示雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視盤邊界欠清,顏色略紅,視網膜血管較細,周邊視網膜色素分布不均,呈椒鹽樣外觀,無色素游離;右眼顳下方視網膜下有黃白色脂性滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血。1B示雙眼光相干斷層掃描像,上圖為右眼,下圖為左眼。右眼以外層結構破壞為主,視網膜色素上皮表面可見顆粒樣強反射,除中心凹處橢圓體帶不連續外,其余部位橢圓體帶消失;左眼中心凹處橢圓體帶結構不清晰,其余部位橢圓體帶消失。1C、1D分別示右眼、左眼熒光素眼底血管造影像。雙眼視盤呈強熒光,深層毛細血管滲漏,周邊視網膜血管滲漏;右眼顳下方血管閉塞,見小片狀無灌注
給予患者右眼后Tenon囊下注射曲安奈德20 mg。患者失訪4個月后復診:視力穩定,眼部檢查無明顯變化。考慮患者病情轉歸與視網膜血管炎的特征不相符,追問病史,患者訴自幼夜盲史。進一步完善相關檢查。眼底自身熒光檢查,雙眼中周部熒光變暗(圖2A)。視野檢查,雙眼明顯向心性縮窄(圖2B)。視網膜電圖檢查,雙眼明、暗視反應均未引出波形。結合患者眼底表現,考慮視網膜色素變性(RP)可能。對患者及其父母外周血行基因檢測(北京中因科技有限公司),結果顯示患者攜帶半合子變異,系RPGR基因c.935-1G>A位點突變(圖3)。其父親、母親均為野生型。最終診斷:雙眼X性連鎖RP(XLRP)合并右眼Coats樣改變。給予患者右眼顳下方視網膜異常血管病灶處行鞏膜外冷凍聯合后Tenon囊下注射曲安奈德20 mg治療,2周后對顳下方補充視網膜激光光凝。治療后4個月復診,右眼、左眼BCVA分別為0.3、0.5;患者右眼顳下方視網膜病灶區血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小(圖4)。隨訪期間曾出現一過性高眼壓,藥物能夠控制。
圖2
Coats樣視網膜色素變性患者復診時眼部檢查像 2A示眼底自身熒光像,左圖為右眼,右圖為左眼。雙眼中周部熒光變暗。2B示視野像,左圖為右眼,右圖為左眼。雙眼視野向心性狹窄
圖3
Coats樣視網膜色素變性患者及其父母基因測序圖 3A示患者,攜帶半合子變異,系RPGR基因c.935-1G>A位點突變;3B示患者父親;3C示患者母親。其父親、母親均為野生型
圖4
Coats樣視網膜色素變性患者治療后右眼彩色眼底像 顳下方視網膜病灶區血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小
討論 RP是一組以光感受器細胞及RPE細胞進行性損害為特征的遺傳性疾病[1]。典型的RP眼底常常表現為視盤蠟黃、視網膜血管一致性狹窄、骨細胞樣色素沉著等,結合病史和其他檢查,通常不容易誤診。但是對于一些不典型的RP,如眼底缺乏骨細胞樣色素沉著,可伴有視網膜血管滲漏、甚至黃斑水腫者[2],可能導致誤診。本例患者玻璃體細胞(+),雙眼視盤顏色略紅,視網膜血管較細,周邊視網膜無明顯的色素游離;FFA檢查示雙眼視盤強熒光,深層毛細血管及周邊視網膜血管熒光素滲漏;有炎癥性表現,未關注到特征性RP的表現,因此首診時誤診為雙眼視網膜血管炎。
Coats樣RP目前文獻報道極少,其在RP中的發病率約1%~5%[3]。該病無明顯性別差異,常雙眼發病,呈對稱性;Coats樣改變通常在RP診斷以后,表現為富含脂性的漿液性視網膜脫離,表面血管扭曲擴張,病灶常位于赤道部或之前,局限于顳下象限,偶可累及后極部黃斑與視盤,且常為多灶性,滲出病變此消彼長;FFA檢查可見RPE點狀透見熒光,視網膜脫離處毛細血管擴張,微動脈瘤,視網膜新生血管形成,晚期熒光素滲漏,周邊無灌注,伴毛細血管擴張滲漏[3]。Coats樣RP常常需要與Coats病鑒別,尤其是眼底表現不典型的RP。Coats病是一種非遺傳性滲出性視網膜血管疾病,好發于年輕男性,單眼發病,主要表現為視網膜毛細血管擴張癥,微動脈瘤,視網膜內、下脂質沉積和纖維化,以及不同程度的滲出性視網膜脫離。根據患者的臨床表現、性別和發病特點,結合遺傳學特征,往往不難鑒別。本例患者自幼夜盲史;雙眼發病,視網膜血管較細,周邊視網膜色素分布不均;視野檢查示雙眼明顯向心性縮窄;ERG檢查示雙眼明、暗視反應均未引出波形;且基因檢測發現RPGR基因c.935-1G>A位點突變。即RP診斷成立。同時,本例患者右眼顳下方視網膜下有黃白色脂質滲出伴視網膜局限性隆起,其表面血管節段性狹窄、擴張,血管壁見白鞘,伴散在微動脈瘤及點片狀出血;再結合其FFA檢查表現,符合RP合并右眼Coats樣改變的診斷。
該病的發病機制可能是由于光感受器和RPE降解產生的有毒物質累積,誘發血管舒張反應,以及視網膜血管狹窄導致視網膜組織慢性缺血、缺氧,從而引發毛細血管擴張、微動脈瘤及視網膜新生血管形成[4-5]。
Coats樣RP的變異基因包括CRB1、RPGR、CRX、CEP290、PRPF8、CNGB1、TULP1等[3, 5],其中以CRB1突變報道較多,被認為是Coats樣RP的重要危險因素。Demirci等[6]報道了1例XLRP合并Coats樣病例,發現RPGR基因外顯子ORF15發生突變(912G>T)。本例患者致病基因為RPGR,c.935-1G>A 位點變異,其父親、母親均為野生型。該位點為新發突變位點,僅在ClinVar數據庫中收錄為致病性,在外顯子組聚集聯盟數據庫、人群基因頻率數據庫、人類基因致病性突變數據庫數據庫均未收錄。
目前Coats樣RP的治療主要針對視網膜異常血管病變導致的并發癥,輕者通常采用視網膜激光光凝、鞏膜外冷凍封閉異常血管病變;玻璃體腔注射抗血管內皮生長因子藥物減輕視網膜出血、滲出;對于伴有嚴重的視網膜脫離可行視網膜下液引流、鞏膜外墊壓,甚至玻璃體切割手術[3]。本例患者經鞏膜外冷凍封閉異常血管病變及后Tenon囊下注射曲安奈德治療,同時后期輔以視網膜激光光凝補充治療后,視網膜血管擴張減輕,出血減少,硬性滲出部分吸收,局限性視網膜脫離范圍縮小。