引用本文: 成芳, 梁建宏. KIF11基因新突變致MCLMR綜合征1例. 中華眼底病雜志, 2023, 39(7): 583-584. doi: 10.3760/cma.j.cn511434-20220425-00246 復制
患兒女,3歲3個月。患兒3月齡因“不追視”于外院診斷為“家族性滲出性視網膜病變(FEVR)?發育落后”,為進一步明確診斷于2018年10月首次就診于北京大學人民醫院眼科門診。患兒為第2胎,第2產,胎齡39周順產,出生體重3 100 g;否認缺氧窒息史及輸血史。母孕早期曾重感冒行口服藥、輸液治療(具體用藥不詳),孕晚期產檢B型超聲檢查提示胎兒雙頂徑小。父母非近親結婚。患兒分別于4月齡、6月齡、10月齡、1歲3月齡、2歲4月齡、3歲3月齡于北京大學人民醫院眼科門診復診。現3歲3個月,患兒不與人交流,低頭,會簡單地說“爸爸、媽媽”,陌生環境不敢獨走。頭圍(枕額徑)45.2 cm(-2 SD),面部寬鼻梁,招風耳,厚下唇,薄上唇。未見下肢及足部淋巴水腫。
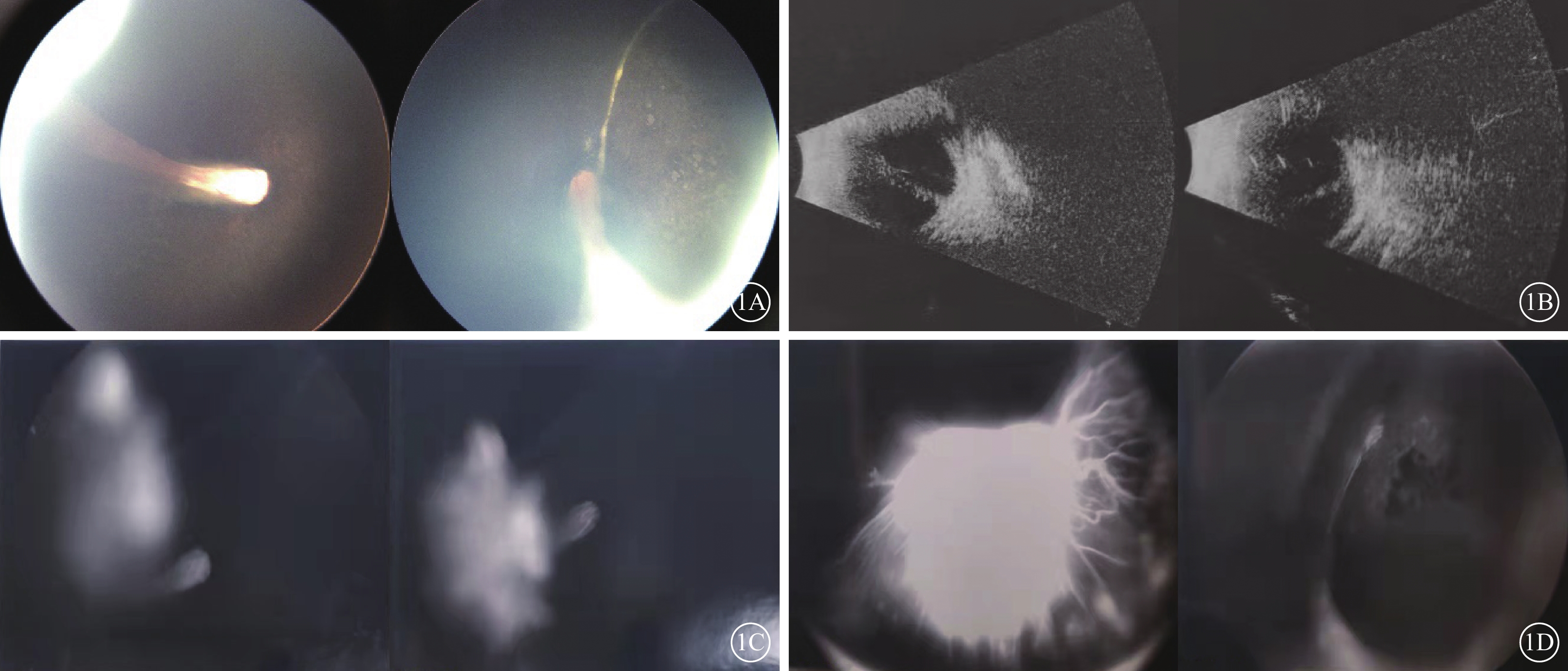
患兒首診至今多次行廣域數碼小兒視網膜成像系統眼底檢查,均示視網膜皺襞(圖1A);B型超聲檢查,雙眼顳下方視網膜鐮狀皺襞(圖1B)。A型超聲測得眼軸右眼、左眼分別為15.9、16.1 mm,提示小眼球。10月齡時散瞳驗光,右眼+2.75 DS/+2.00 DC×155°,左眼+1.75DS/+2.75 DC×160°,表現為復合遠視散光。1歲3月齡時行熒光素眼底血管造影檢查,雙眼自視盤起巨大視網膜皺襞,其上布滿血管,呈強熒光,延至顳側眼底周邊及晶狀體后,后極部未見血管(圖1C,1D)。患兒多次復診眼前節及眼壓未見異常,眼底表現未見明顯變化。結合患兒3月齡時頭顱、眼眶核磁共振檢查結果“頭顱較小,雙側額顳部蛛網膜下腔略寬,白質髓鞘化落后于1月齡兒;雙側眼球小,玻璃體內自晶狀體向后可見線狀短T2信號分隔”,臨床診斷:MCLMR綜合征。
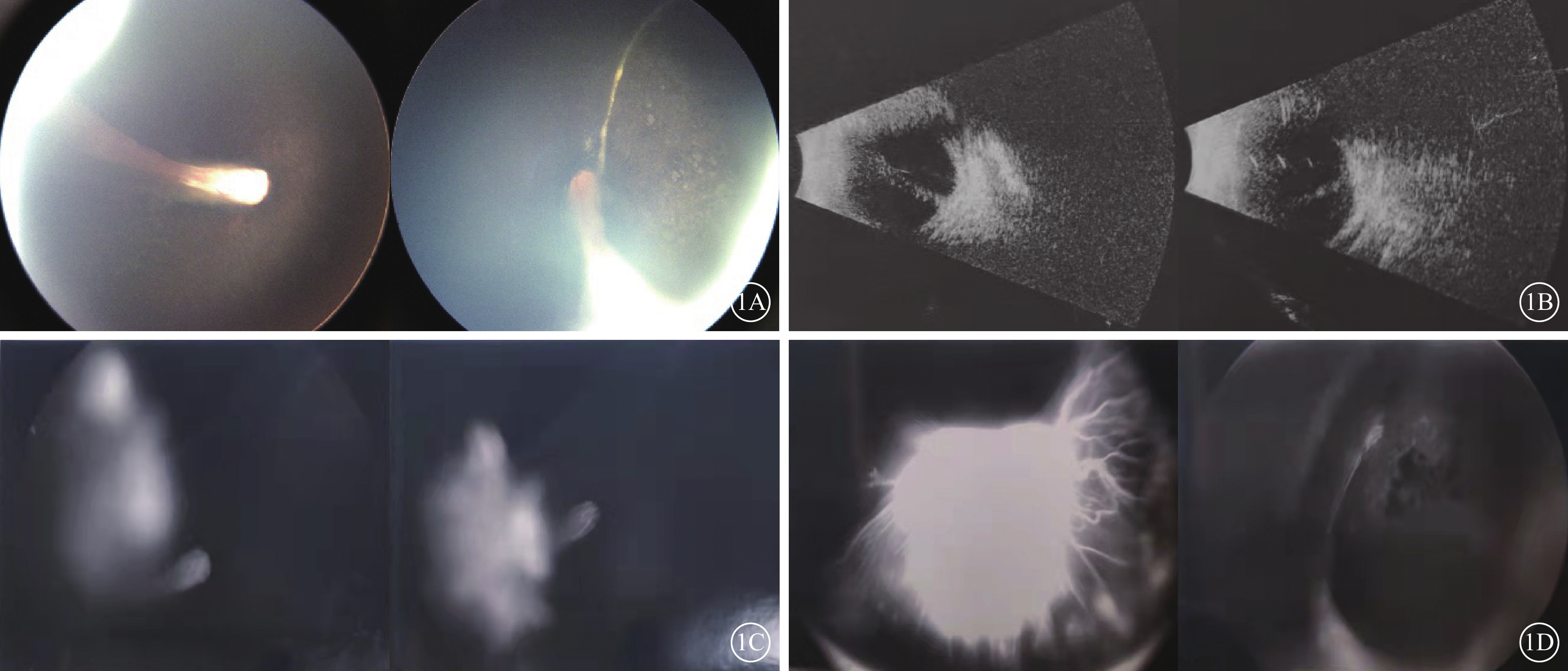 圖1
KIF11基因新突變致MCLMR綜合征患兒眼部檢查像 1A示患兒3月齡雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視網膜皺襞。1B示患兒3月齡眼B型超聲像,左圖為右眼,右圖為左眼。雙眼顳下方視網膜鐮狀皺襞。1C、1D分別示患兒1歲3月齡右眼、左眼熒光素眼底血管造影像,右眼自視盤起視網膜皺襞;左眼自視盤起巨大視網膜皺襞,后極部無血管
圖1
KIF11基因新突變致MCLMR綜合征患兒眼部檢查像 1A示患兒3月齡雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視網膜皺襞。1B示患兒3月齡眼B型超聲像,左圖為右眼,右圖為左眼。雙眼顳下方視網膜鐮狀皺襞。1C、1D分別示患兒1歲3月齡右眼、左眼熒光素眼底血管造影像,右眼自視盤起視網膜皺襞;左眼自視盤起巨大視網膜皺襞,后極部無血管
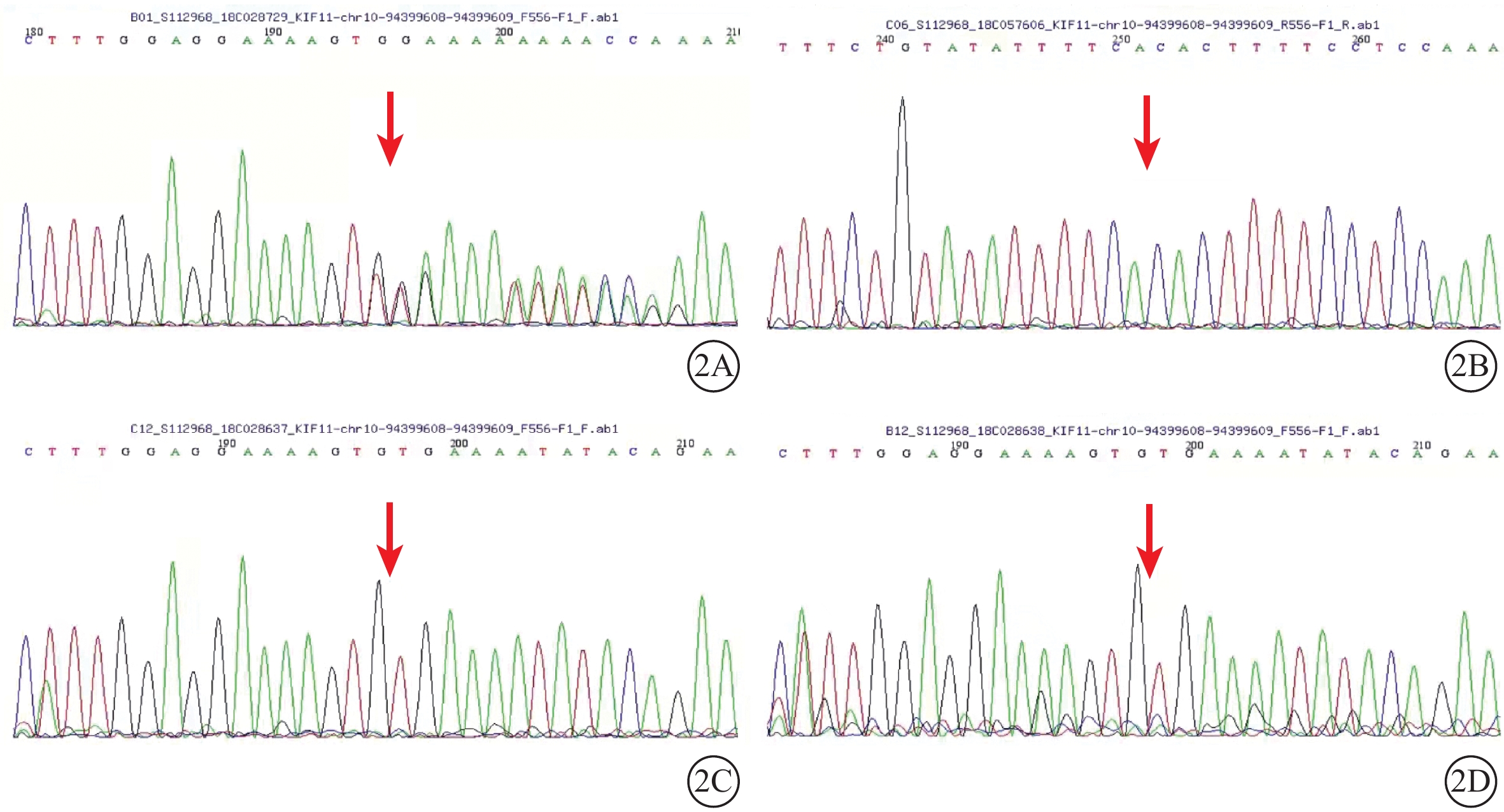
采集患兒及其父母、姐姐外周靜脈血行基因檢測。對患兒采用全外顯子組測序,檢測結果通過生物信息學分析后得到候選致病突變位點。運用Sanger測序進行驗證及家系共分離分析,確定致病性突變位點。檢測結果顯示,患兒存在KIF11基因雜合移碼突變c.2219delG(p.C740Lfs*21)(圖2A),蛋白功能預測軟件REVEL鑒定該變異位點預測致病性為未知;根據美國醫學遺傳學和基因組學推薦指南對該變異位點進行致病等級評估為致病性變異。其父母、姐姐均未攜帶該位點基因突變(圖2B~2D),提示患兒KIF11基因突變為自發突變。KIF11基因轉錄本編號NM_0045。
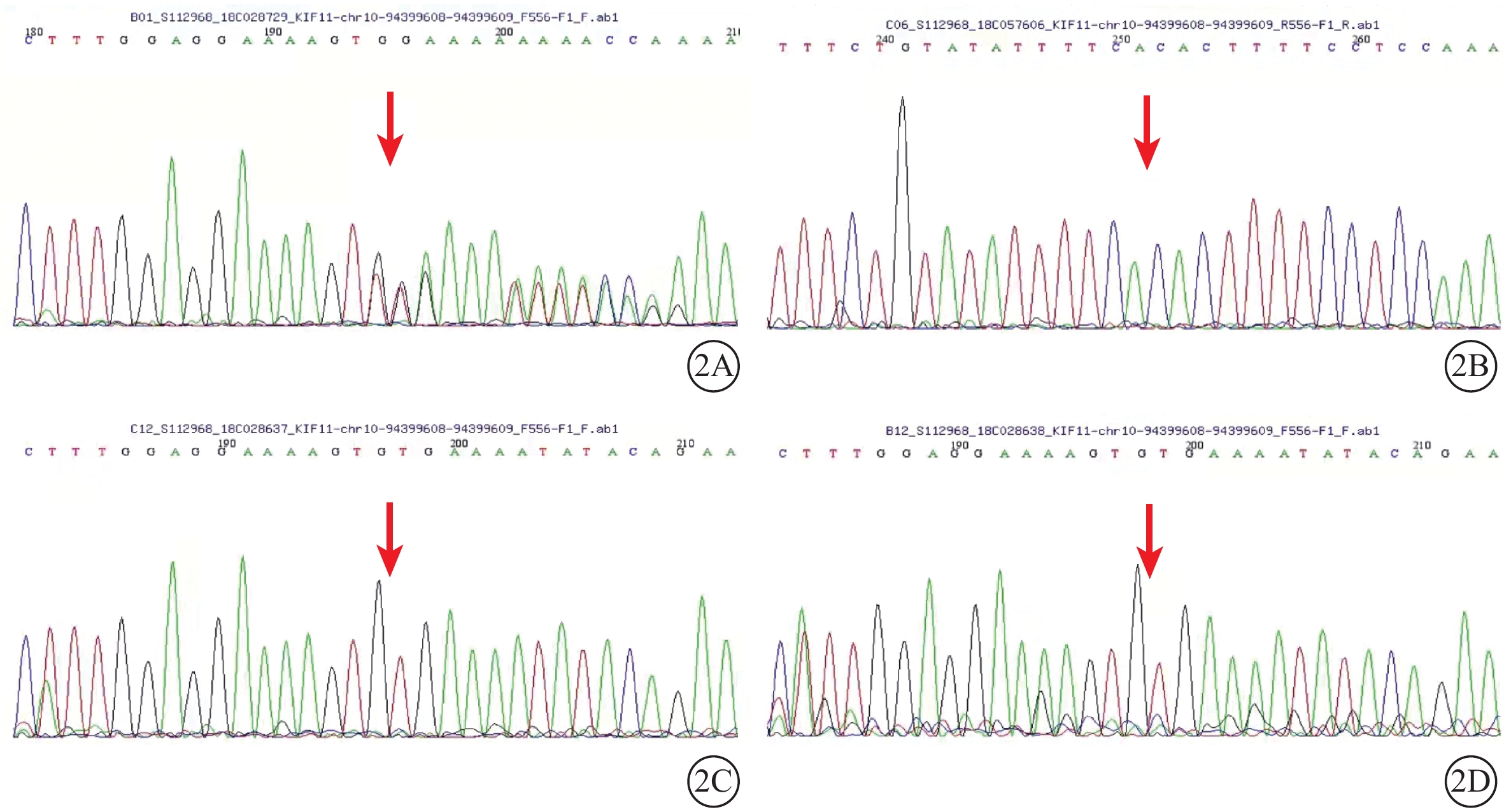 圖2
KIF11基因新突變致MCLMR綜合征患兒及其親屬基因測序圖 2A示患兒,其存在KIF11基因c.2219delG(p.C740Lfs*21)雜合移碼突變(紅箭);2B~2D分別示患兒父親、姐姐、母親,該位點均不存在變異(紅箭)
圖2
KIF11基因新突變致MCLMR綜合征患兒及其親屬基因測序圖 2A示患兒,其存在KIF11基因c.2219delG(p.C740Lfs*21)雜合移碼突變(紅箭);2B~2D分別示患兒父親、姐姐、母親,該位點均不存在變異(紅箭)
討論 MCLMR綜合征是一種罕見的遺傳性疾病,以小頭畸形伴或不伴脈絡膜視網膜病變、淋巴水腫或智力遲緩為主要特征,臨床表現變異大。Opitz[1]于1986年首次將面部特征(如鼻梁寬、凹陷、鼻尖突出、嘴唇豐滿、輕度小頜畸形)描述為面部先天性淋巴水腫。既往文獻報道的臨床資料完整的89例患者中,以小頭畸形、眼部異常最常見,尤其是脈絡膜視網膜病變,其次為智力發育落后和淋巴水腫。本例患兒胚胎期、3月齡及3歲時均表現為小頭癥,眼部異常主要表現為小眼球、復合遠視散光、視網膜皺襞,智力輕度發育落后,新生兒期未見明顯下肢及足背淋巴水腫,但表現出特殊面部形態。其表現出典型的MCLMR綜合征臨床表現。
2012年,Ostergaard等[2]首次鑒定了KIF11基因中的突變可導致MCLMR綜合征,該基因突變以常染色體顯性方式遺傳。隨后陸續有KIF11基因突變導致MCLMR綜合征的報道[2-11]。本例患者KIF11 p.C740Lfs*21變異既往未見報道,為新突變。患兒父母及姐姐未攜帶KIF11基因突變,該患兒屬自發突變。
2014年Robitaille等[5]在FEVR患者中鑒定出KIF11基因突變,表明FEVR和MCLMR綜合征之間存在表型重疊。雖然MCLMR的眼部特征與FEVR類似,但對于眼科首診的攜帶KIF11基因致病突變的視網膜皺襞患者,我們還需要關注其是否有小頭畸形、淋巴水腫及智力低下等全身表現,以明確FEVR或MCLMR綜合征診斷。
患兒女,3歲3個月。患兒3月齡因“不追視”于外院診斷為“家族性滲出性視網膜病變(FEVR)?發育落后”,為進一步明確診斷于2018年10月首次就診于北京大學人民醫院眼科門診。患兒為第2胎,第2產,胎齡39周順產,出生體重3 100 g;否認缺氧窒息史及輸血史。母孕早期曾重感冒行口服藥、輸液治療(具體用藥不詳),孕晚期產檢B型超聲檢查提示胎兒雙頂徑小。父母非近親結婚。患兒分別于4月齡、6月齡、10月齡、1歲3月齡、2歲4月齡、3歲3月齡于北京大學人民醫院眼科門診復診。現3歲3個月,患兒不與人交流,低頭,會簡單地說“爸爸、媽媽”,陌生環境不敢獨走。頭圍(枕額徑)45.2 cm(-2 SD),面部寬鼻梁,招風耳,厚下唇,薄上唇。未見下肢及足部淋巴水腫。
患兒首診至今多次行廣域數碼小兒視網膜成像系統眼底檢查,均示視網膜皺襞(圖1A);B型超聲檢查,雙眼顳下方視網膜鐮狀皺襞(圖1B)。A型超聲測得眼軸右眼、左眼分別為15.9、16.1 mm,提示小眼球。10月齡時散瞳驗光,右眼+2.75 DS/+2.00 DC×155°,左眼+1.75DS/+2.75 DC×160°,表現為復合遠視散光。1歲3月齡時行熒光素眼底血管造影檢查,雙眼自視盤起巨大視網膜皺襞,其上布滿血管,呈強熒光,延至顳側眼底周邊及晶狀體后,后極部未見血管(圖1C,1D)。患兒多次復診眼前節及眼壓未見異常,眼底表現未見明顯變化。結合患兒3月齡時頭顱、眼眶核磁共振檢查結果“頭顱較小,雙側額顳部蛛網膜下腔略寬,白質髓鞘化落后于1月齡兒;雙側眼球小,玻璃體內自晶狀體向后可見線狀短T2信號分隔”,臨床診斷:MCLMR綜合征。
圖1
KIF11基因新突變致MCLMR綜合征患兒眼部檢查像 1A示患兒3月齡雙眼彩色眼底像,左圖為右眼,右圖為左眼。雙眼視網膜皺襞。1B示患兒3月齡眼B型超聲像,左圖為右眼,右圖為左眼。雙眼顳下方視網膜鐮狀皺襞。1C、1D分別示患兒1歲3月齡右眼、左眼熒光素眼底血管造影像,右眼自視盤起視網膜皺襞;左眼自視盤起巨大視網膜皺襞,后極部無血管
采集患兒及其父母、姐姐外周靜脈血行基因檢測。對患兒采用全外顯子組測序,檢測結果通過生物信息學分析后得到候選致病突變位點。運用Sanger測序進行驗證及家系共分離分析,確定致病性突變位點。檢測結果顯示,患兒存在KIF11基因雜合移碼突變c.2219delG(p.C740Lfs*21)(圖2A),蛋白功能預測軟件REVEL鑒定該變異位點預測致病性為未知;根據美國醫學遺傳學和基因組學推薦指南對該變異位點進行致病等級評估為致病性變異。其父母、姐姐均未攜帶該位點基因突變(圖2B~2D),提示患兒KIF11基因突變為自發突變。KIF11基因轉錄本編號NM_0045。
圖2
KIF11基因新突變致MCLMR綜合征患兒及其親屬基因測序圖 2A示患兒,其存在KIF11基因c.2219delG(p.C740Lfs*21)雜合移碼突變(紅箭);2B~2D分別示患兒父親、姐姐、母親,該位點均不存在變異(紅箭)
討論 MCLMR綜合征是一種罕見的遺傳性疾病,以小頭畸形伴或不伴脈絡膜視網膜病變、淋巴水腫或智力遲緩為主要特征,臨床表現變異大。Opitz[1]于1986年首次將面部特征(如鼻梁寬、凹陷、鼻尖突出、嘴唇豐滿、輕度小頜畸形)描述為面部先天性淋巴水腫。既往文獻報道的臨床資料完整的89例患者中,以小頭畸形、眼部異常最常見,尤其是脈絡膜視網膜病變,其次為智力發育落后和淋巴水腫。本例患兒胚胎期、3月齡及3歲時均表現為小頭癥,眼部異常主要表現為小眼球、復合遠視散光、視網膜皺襞,智力輕度發育落后,新生兒期未見明顯下肢及足背淋巴水腫,但表現出特殊面部形態。其表現出典型的MCLMR綜合征臨床表現。
2012年,Ostergaard等[2]首次鑒定了KIF11基因中的突變可導致MCLMR綜合征,該基因突變以常染色體顯性方式遺傳。隨后陸續有KIF11基因突變導致MCLMR綜合征的報道[2-11]。本例患者KIF11 p.C740Lfs*21變異既往未見報道,為新突變。患兒父母及姐姐未攜帶KIF11基因突變,該患兒屬自發突變。
2014年Robitaille等[5]在FEVR患者中鑒定出KIF11基因突變,表明FEVR和MCLMR綜合征之間存在表型重疊。雖然MCLMR的眼部特征與FEVR類似,但對于眼科首診的攜帶KIF11基因致病突變的視網膜皺襞患者,我們還需要關注其是否有小頭畸形、淋巴水腫及智力低下等全身表現,以明確FEVR或MCLMR綜合征診斷。