引用本文: 杜婉麗, 吳彩云, 連露露, 張釧, 王玉佩, 郝勝菊, 惠玲, 張慶華. NDP基因變異一家系的臨床特征及基因分析和產前診斷. 中華眼底病雜志, 2023, 39(7): 549-553. doi: 10.3760/cma.j.cn511434-20221008-00527 復制
Norrin蛋白是一種由NDP基因編碼的富含半胱氨酸的分泌蛋白,在視網膜血管發育中起著核心作用,對視網膜的分化和維持至關重要[1]。NDP基因變異可導致 Norrie病(ND,MIM310600),極少數情況下還會導致家族性滲出性玻璃體視網膜病變(FEVR,MIM305390)和早產兒視網膜病變(ROP)[2]。截止目前,人類基因變異數據庫(HGMD)已經收錄200多個NDP基因突變,不同變異可導致臨床表型不同,且ND、FEVR、ROP在臨床體征上也有交叉,往往導致臨床不能明確診斷。我們通過1個NDP基因變異家系及家系不同成員的臨床表型觀察,以期為此類基因突變的研究提供依據。現將結果報道如下。
1 對象和方法
家系調查研究。本研究經甘肅省婦幼保健院倫理委員會批準(批文號:2021-24KYSL); 遵循《赫爾辛基宣言》原則;患兒監護人及家系成員均簽署書面知情同意書。
2019年7月至2021年12月于甘肅省婦幼保健院眼科就診的NDP基因突變的一個3代漢族家系的2例患者和6名家系成員(圖1)納入本研究。先證者由母親提供外院檢查病歷,提示雙眼視網膜全脫離。先證者父母及2個弟弟接受詳細眼科專科檢查,包括瞳孔對光反射、帶狀光檢影、最佳矯正視力、追視視力評估、眼底彩色照相、廣角熒光素眼底血管造影(FFA)檢查。詳細詢問并記錄家族史、婚育史及其他全身疾病史。
 圖1
NDP基因突變家系圖 ↗:先證者;■:男性患者;
圖1
NDP基因突變家系圖 ↗:先證者;■:男性患者; :女性攜帶者;□:正常男性;○:正常女性
:女性攜帶者;□:正常男性;○:正常女性
采集受檢者外周靜脈血5 ml,乙二胺四乙酸抗凝。采用天根生化科技(北京)有限公司的全血液基因組DNA提取試劑盒按照操作規程提取全基因組DNA,Nanodrop2000核酸定量儀測定其純度和濃度,符合要求的基因組DNA分裝后保存于-20 ℃待用。
全外顯子組測序(WES)、多重連接探針擴增(MLPA)檢測。采用GenCap液相捕獲試劑盒及標準文庫構建試劑盒(邁基諾公司自主研發)對DNA目標區域進行捕獲,構建文庫。采用美國Illumina Nextseq 500高通量測序儀對DNA樣本進行測序,篩選致病基因。對候選變異位點進行MLPA驗證。MLPA檢測NDP基因。采用荷蘭MRC-Holland公司SALSA MLPA 探針組 P0285試劑盒按照說明書操作。采用美國ABI公司3500 DX測序儀對反應產物進行毛細管電泳。應用Coffalyser軟件對結果進行分析。
先證者母親于再次妊娠19周時,超聲引導下行羊膜腔穿刺,抽取羊水15 ml。采用天根生化科技(北京)有限公司的羊水細胞基因組DNA提取試劑盒按照操作規程提取基因組DNA,-20 ℃保存備用。胎兒DNA母體污染排除:采用美國ABI公司3500 DX測序儀分別對胎兒DNA及胎兒父母外周血的16個短串聯重復序列(STR)位點進行測定。胎兒STR位點等位基因的熒光峰分別來自父母,胎兒STR位點無來自母親的2個熒光檢測峰即可排除母體污染后,行NDP基因MLPA檢測,步驟同前。
2 結果
先證者(Ⅲ1),男,4歲,足月順產。出生40 d左右不追視,外院B型超聲檢查提示雙眼視網膜全脫離。聽力、智力檢查均正常。未能配合行眼底檢查。先證者母親(Ⅱ2),28歲。雙眼視力1.0;眼壓正常。眼底檢查,顳側周邊視網膜未血管化;廣角FFA檢查,顳側周邊視網膜未血管化,末梢血管熒光素輕度滲漏(圖2A)。臨床診斷:雙眼周邊視網膜血管異常。先證者第1同胞(Ⅲ2,大弟),出生30 d于我院行眼科檢查。雙眼角膜、晶狀體透明;眼壓正常;雙眼視網膜脫離(圖2B,2C)。先證者第2同胞(Ⅲ3,小弟),其母孕周19產前診斷提示為男胎,出生后1 d于我院行新生兒眼病篩查。雙眼外眼未見異常;角膜、晶狀體透明;雙眼視盤、黃斑未見異常,周邊視網膜血管化,未見血管紆曲(圖2D)。先證者父親(Ⅱ1),26歲,雙眼祼眼視力1.0;眼壓、眼底彩色照相、廣角FFA檢查均未見異常。
 圖2
NDP基因突變家系受檢者眼部檢查像 2A示先證者母親(Ⅱ2)熒光素眼底血管造影像,顳側周邊視網膜未見熒光充盈,少量熒光素滲漏;2B、2C分別示先證者大弟(Ⅲ2)右眼、左眼彩色眼底像,雙眼視網膜脫離,視網膜結構不清;2D示先證者小弟(Ⅲ3)彩色眼底像,視網膜未見異常,雙眼視網膜血管發育至周邊視網膜
圖2
NDP基因突變家系受檢者眼部檢查像 2A示先證者母親(Ⅱ2)熒光素眼底血管造影像,顳側周邊視網膜未見熒光充盈,少量熒光素滲漏;2B、2C分別示先證者大弟(Ⅲ2)右眼、左眼彩色眼底像,雙眼視網膜脫離,視網膜結構不清;2D示先證者小弟(Ⅲ3)彩色眼底像,視網膜未見異常,雙眼視網膜血管發育至周邊視網膜
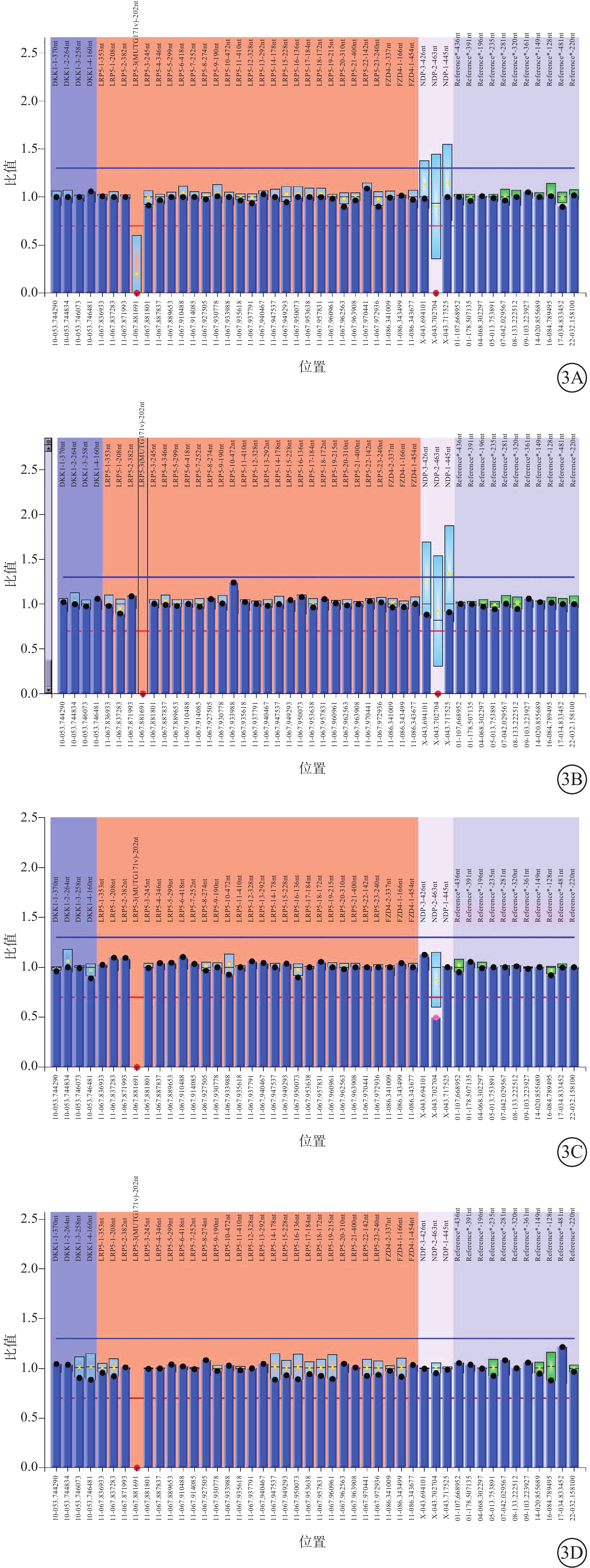
WES結果顯示,該家系chrX-43817718-43818168區域存在半合子缺失(該區域包括NDP基因第2號外顯子)。先證者母親為雜合缺失;父親未見變異。MLPA檢測驗證結果顯示,先證者(Ⅲ1)、其大弟(Ⅲ2)NDP基因的第2號外顯子存在半合子缺失突變(圖3A,3B);其母親(Ⅱ2)NDP基因第2號外顯子存在缺失雜合突變(圖3C)。該變異在HGMD及相關文獻已有收錄和報道[3-4],為致病性變異,與ND和FEVR相關。
 圖3
NDP基因突變家系受檢者多重連接探針擴增檢測圖 3A、3B分別示先證者(Ⅲ1)及大弟(Ⅲ2)NDP基因第2號外顯子存在半合子缺失突變;3C示先證者母親(Ⅱ2),NDP基因第2號外顯子存在雜合缺失突變;3D示先證者小弟“胎兒”(Ⅲ3),未檢測到該變異
圖3
NDP基因突變家系受檢者多重連接探針擴增檢測圖 3A、3B分別示先證者(Ⅲ1)及大弟(Ⅲ2)NDP基因第2號外顯子存在半合子缺失突變;3C示先證者母親(Ⅱ2),NDP基因第2號外顯子存在雜合缺失突變;3D示先證者小弟“胎兒”(Ⅲ3),未檢測到該變異
先證者母親再次生育時產前診斷結果,胎兒羊水MLPA檢測提示胎兒SRY(+);未檢測到與先證者相同的NDP基因第2號外顯子缺失變異(圖3D)。
3 討論
NDP基因位于X染色體短臂(p11.4),其產物Norrin蛋白可作為配體激活Wnt/β-catenin信號通路,調節血管生成、維持血視網膜屏障和血腦屏障穩態等功能[5-6]。而該通路異常可導致一系列的玻璃體視網膜疾病,HGMD目前共收錄了200余個NDP基因突變,患者主要表現為ND表型,僅5%的患者表現為FEVR。
ND和FEVR有不同的臨床表型,但部分表型存在交叉,因此臨床診斷并不容易。ND患兒出生時或嬰幼兒時期內表現為白瞳和視力喪失,通常雙側對稱發病,眼底表現為灰色或灰黃色假神經膠質瘤樣(“南瓜樣”)視網膜發育不良,晶狀體后纖維增生,視網膜皺褶及視網膜脫離[7];其他還常見感覺性聽力喪失和認知障礙,以及行為障礙、癲癇發作和周圍血管病變[8]。FEVR臨床表現主要為不同階段的視網膜血管異常和病變,如視網膜周邊無血管區、視網膜血管走形異常、玻璃體視網膜牽引、視網膜劈裂從視盤延伸至顳側周邊、牽拉性視網膜脫離[9]。因此,ND眼部表現較典型FEVR更嚴重和發育不良,通常在出生后3個月內被檢測到,而FEVR則在不同的階段被檢測到。臨床ND與FEVR的鑒別診斷極其具有挑戰性,ND通常與兒童早期的智力殘疾或進行性感音神經性聽力損失有關[10]。
本家系中患者(先證者及其大弟)基因型為NDP基因半合子變異,臨床表現均為出生后即全視網膜脫離,其中大弟眼底表現為后極部視網膜呈團塊樣,團塊中央疑似有黃色滲出樣組織,類似假性膠質瘤的表現,符合NDP基因相關的ND眼底表現。先證者母親為NDP基因雜合變異攜帶者,眼底表現為顳側周邊視網膜無血管區,無視力、聽力、智力及精神異常,結合臨床表現,考慮為NDP基因相關的FEVR。有研究發現,12個攜帶NDP致病基因變異的女性攜帶者多表現出FEVR的臨床特征,其中第2號外顯子缺失病例,臨床表現與本家系病例相似[4]。另有文獻報道,NDP基因拷貝數變異所導致的3例患者均為男性,且均在出生后不久發現視網膜脫離[11],但此研究未對此表型做出臨床診斷。 因此,在本家系中,我們驗證了既往文獻中NDP基因拷貝數變異在不同性別中的臨床表現。
對于本家系臨床診斷是ND還是FEVR,我們想追蹤家系中其他成員是否存在有聽力和智力異常,再進一步確診,但先證者母親不愿做臨床評估。本家系確診病例中,臨床表現不能明確是哪一種疾病,我們考慮是否可從第2號外顯子的缺失,影響蛋白質功能方面分析其臨床表型。
NDP基因跨越28 kb基因組,包含3個外顯子,編碼部分跨越第2號外顯子的后半部分和第3號外顯子的第一部分,開放閱讀框的前58個殘基位于第2號外顯子,而開放閱讀框的密碼子59~133和3'非翻譯區位于第3號外顯子[12]。Norrie的胱氨酸結結構域從32~133密碼子,被認為在神經相互作用中發揮重要作用[13]。在密碼子39、65、69、96、126、128處的半胱氨酸殘基被發現負責半胱氨酸結的形成。密碼子39和96、65和126與密碼子69和128之間的3個二硫橋參與了Norrin的三級結構[1]。
Wu等[1]分析了5例ND患者和4例FEVR患者的NDP基因變異,發現ND患者的變異涉及Norrie蛋白的半胱氨酸殘基,而Norrie蛋白的非半胱氨酸殘基突變顯示血管和視網膜發育異常,更符合X連鎖遺傳的FEVR。破壞半胱氨酸結基序的突變與嚴重視網膜發育不良相對應,而非半胱氨酸突變患者有不同程度的無血管周圍視網膜、視網膜外血管系統和視網膜下滲出物。部分NDP基因研究表明,潛在連續缺失較基因內變異(小變異或第2號外顯子缺失)的患者更有可能出現認知障礙和行為表型[6],基因內微結構的缺失更容易引起神經癥狀[14]。
本家系患者的NDP基因第2號外顯子缺失,缺失部位涉及半胱氨酸結構,為ND表現,且家系中應有聽力和神經癥狀表現者,但2例男性患兒表現為嚴重的視網膜脫離和出生后盲,母親無視力異常表現,僅在FFA檢查中發現周邊視網膜的無血管區。既往文獻對于第2號外顯子缺失的報道較少,檢索到的文獻也是視力喪失患者的基因檢測和臨床表現,無家系的臨床表現追蹤[4, 10]。在HGMD中收錄有1例NDP基因第2號外顯子缺失的報道[15],此文獻是對于患者聽力的20年追蹤觀察,患者出生時視力喪失,在青春期才出現感覺性聽力損失。
對于NDP基因突變家系中臨床表現不同的研究,Liu等[16]曾報道1例NDP基因變異所致男性ND患兒。患兒2歲后出現學習和與他人交流方面的困難,同時出現指壓眼征和自傷行為;4歲時因雙側白內障及視力差就診,B型超聲檢查顯示雙眼視網膜脫離。患兒父親有與脊髓灰質炎相關的身體殘疾,眼部檢查未見異常;母親左眼有眼球癆和角膜病,視力無光感;姨媽雙眼眼底檢查正常;基因檢測母親和姨媽NDP基因中存在第2、3號外顯子的純合子缺失,診斷為ND。
因此,本家系也許是ND在家系中雜合和純合的表現型不同,或許就是FEVR,亦或是兩種疾病在表現上的重合。或與部分研究者意見一致,NDP基因突變涉及廣泛的Wnt信號異常,因而FEVR和ND并非總能做出明確診斷[17]。我們將繼續追蹤觀察家系中其他成員的表現,患者聽力、神經以及其他眼外表現,也許多年后才能最終確定是ND或FEVR。
本家系中先證者母親在第三胎懷孕19周后接受了羊膜穿刺基因檢測,未發現胎兒基因突變。妊娠30周時,對胎兒眼部進行了超聲檢查,顯示對稱球狀,透明明亮的晶狀體和玻璃體腔。出生后眼科檢查符合無NDP基因突變表現。本研究結果證實,在遺傳性視網膜疾病家庭中,羊膜穿刺產前診斷是有效阻斷家族遺傳疾病的方式,為懷孕一個健康的孩子提供安全、可靠的幫助。
NDP基因第2號外顯子缺失的臨床表型在一個家系有兩種表現,對于此家系我們還將進行長期隨訪,觀察眼部體征的發展情況,以及眼外的其他體征,為NDP基因突變的基因型和表型的關系研究提供依據。
Norrin蛋白是一種由NDP基因編碼的富含半胱氨酸的分泌蛋白,在視網膜血管發育中起著核心作用,對視網膜的分化和維持至關重要[1]。NDP基因變異可導致 Norrie病(ND,MIM310600),極少數情況下還會導致家族性滲出性玻璃體視網膜病變(FEVR,MIM305390)和早產兒視網膜病變(ROP)[2]。截止目前,人類基因變異數據庫(HGMD)已經收錄200多個NDP基因突變,不同變異可導致臨床表型不同,且ND、FEVR、ROP在臨床體征上也有交叉,往往導致臨床不能明確診斷。我們通過1個NDP基因變異家系及家系不同成員的臨床表型觀察,以期為此類基因突變的研究提供依據。現將結果報道如下。
1 對象和方法
家系調查研究。本研究經甘肅省婦幼保健院倫理委員會批準(批文號:2021-24KYSL); 遵循《赫爾辛基宣言》原則;患兒監護人及家系成員均簽署書面知情同意書。
2019年7月至2021年12月于甘肅省婦幼保健院眼科就診的NDP基因突變的一個3代漢族家系的2例患者和6名家系成員(圖1)納入本研究。先證者由母親提供外院檢查病歷,提示雙眼視網膜全脫離。先證者父母及2個弟弟接受詳細眼科專科檢查,包括瞳孔對光反射、帶狀光檢影、最佳矯正視力、追視視力評估、眼底彩色照相、廣角熒光素眼底血管造影(FFA)檢查。詳細詢問并記錄家族史、婚育史及其他全身疾病史。
圖1
NDP基因突變家系圖 ↗:先證者;■:男性患者;:女性攜帶者;□:正常男性;○:正常女性
采集受檢者外周靜脈血5 ml,乙二胺四乙酸抗凝。采用天根生化科技(北京)有限公司的全血液基因組DNA提取試劑盒按照操作規程提取全基因組DNA,Nanodrop2000核酸定量儀測定其純度和濃度,符合要求的基因組DNA分裝后保存于-20 ℃待用。
全外顯子組測序(WES)、多重連接探針擴增(MLPA)檢測。采用GenCap液相捕獲試劑盒及標準文庫構建試劑盒(邁基諾公司自主研發)對DNA目標區域進行捕獲,構建文庫。采用美國Illumina Nextseq 500高通量測序儀對DNA樣本進行測序,篩選致病基因。對候選變異位點進行MLPA驗證。MLPA檢測NDP基因。采用荷蘭MRC-Holland公司SALSA MLPA 探針組 P0285試劑盒按照說明書操作。采用美國ABI公司3500 DX測序儀對反應產物進行毛細管電泳。應用Coffalyser軟件對結果進行分析。
先證者母親于再次妊娠19周時,超聲引導下行羊膜腔穿刺,抽取羊水15 ml。采用天根生化科技(北京)有限公司的羊水細胞基因組DNA提取試劑盒按照操作規程提取基因組DNA,-20 ℃保存備用。胎兒DNA母體污染排除:采用美國ABI公司3500 DX測序儀分別對胎兒DNA及胎兒父母外周血的16個短串聯重復序列(STR)位點進行測定。胎兒STR位點等位基因的熒光峰分別來自父母,胎兒STR位點無來自母親的2個熒光檢測峰即可排除母體污染后,行NDP基因MLPA檢測,步驟同前。
2 結果
先證者(Ⅲ1),男,4歲,足月順產。出生40 d左右不追視,外院B型超聲檢查提示雙眼視網膜全脫離。聽力、智力檢查均正常。未能配合行眼底檢查。先證者母親(Ⅱ2),28歲。雙眼視力1.0;眼壓正常。眼底檢查,顳側周邊視網膜未血管化;廣角FFA檢查,顳側周邊視網膜未血管化,末梢血管熒光素輕度滲漏(圖2A)。臨床診斷:雙眼周邊視網膜血管異常。先證者第1同胞(Ⅲ2,大弟),出生30 d于我院行眼科檢查。雙眼角膜、晶狀體透明;眼壓正常;雙眼視網膜脫離(圖2B,2C)。先證者第2同胞(Ⅲ3,小弟),其母孕周19產前診斷提示為男胎,出生后1 d于我院行新生兒眼病篩查。雙眼外眼未見異常;角膜、晶狀體透明;雙眼視盤、黃斑未見異常,周邊視網膜血管化,未見血管紆曲(圖2D)。先證者父親(Ⅱ1),26歲,雙眼祼眼視力1.0;眼壓、眼底彩色照相、廣角FFA檢查均未見異常。
圖2
NDP基因突變家系受檢者眼部檢查像 2A示先證者母親(Ⅱ2)熒光素眼底血管造影像,顳側周邊視網膜未見熒光充盈,少量熒光素滲漏;2B、2C分別示先證者大弟(Ⅲ2)右眼、左眼彩色眼底像,雙眼視網膜脫離,視網膜結構不清;2D示先證者小弟(Ⅲ3)彩色眼底像,視網膜未見異常,雙眼視網膜血管發育至周邊視網膜
WES結果顯示,該家系chrX-43817718-43818168區域存在半合子缺失(該區域包括NDP基因第2號外顯子)。先證者母親為雜合缺失;父親未見變異。MLPA檢測驗證結果顯示,先證者(Ⅲ1)、其大弟(Ⅲ2)NDP基因的第2號外顯子存在半合子缺失突變(圖3A,3B);其母親(Ⅱ2)NDP基因第2號外顯子存在缺失雜合突變(圖3C)。該變異在HGMD及相關文獻已有收錄和報道[3-4],為致病性變異,與ND和FEVR相關。
圖3
NDP基因突變家系受檢者多重連接探針擴增檢測圖 3A、3B分別示先證者(Ⅲ1)及大弟(Ⅲ2)NDP基因第2號外顯子存在半合子缺失突變;3C示先證者母親(Ⅱ2),NDP基因第2號外顯子存在雜合缺失突變;3D示先證者小弟“胎兒”(Ⅲ3),未檢測到該變異
先證者母親再次生育時產前診斷結果,胎兒羊水MLPA檢測提示胎兒SRY(+);未檢測到與先證者相同的NDP基因第2號外顯子缺失變異(圖3D)。
3 討論
NDP基因位于X染色體短臂(p11.4),其產物Norrin蛋白可作為配體激活Wnt/β-catenin信號通路,調節血管生成、維持血視網膜屏障和血腦屏障穩態等功能[5-6]。而該通路異常可導致一系列的玻璃體視網膜疾病,HGMD目前共收錄了200余個NDP基因突變,患者主要表現為ND表型,僅5%的患者表現為FEVR。
ND和FEVR有不同的臨床表型,但部分表型存在交叉,因此臨床診斷并不容易。ND患兒出生時或嬰幼兒時期內表現為白瞳和視力喪失,通常雙側對稱發病,眼底表現為灰色或灰黃色假神經膠質瘤樣(“南瓜樣”)視網膜發育不良,晶狀體后纖維增生,視網膜皺褶及視網膜脫離[7];其他還常見感覺性聽力喪失和認知障礙,以及行為障礙、癲癇發作和周圍血管病變[8]。FEVR臨床表現主要為不同階段的視網膜血管異常和病變,如視網膜周邊無血管區、視網膜血管走形異常、玻璃體視網膜牽引、視網膜劈裂從視盤延伸至顳側周邊、牽拉性視網膜脫離[9]。因此,ND眼部表現較典型FEVR更嚴重和發育不良,通常在出生后3個月內被檢測到,而FEVR則在不同的階段被檢測到。臨床ND與FEVR的鑒別診斷極其具有挑戰性,ND通常與兒童早期的智力殘疾或進行性感音神經性聽力損失有關[10]。
本家系中患者(先證者及其大弟)基因型為NDP基因半合子變異,臨床表現均為出生后即全視網膜脫離,其中大弟眼底表現為后極部視網膜呈團塊樣,團塊中央疑似有黃色滲出樣組織,類似假性膠質瘤的表現,符合NDP基因相關的ND眼底表現。先證者母親為NDP基因雜合變異攜帶者,眼底表現為顳側周邊視網膜無血管區,無視力、聽力、智力及精神異常,結合臨床表現,考慮為NDP基因相關的FEVR。有研究發現,12個攜帶NDP致病基因變異的女性攜帶者多表現出FEVR的臨床特征,其中第2號外顯子缺失病例,臨床表現與本家系病例相似[4]。另有文獻報道,NDP基因拷貝數變異所導致的3例患者均為男性,且均在出生后不久發現視網膜脫離[11],但此研究未對此表型做出臨床診斷。 因此,在本家系中,我們驗證了既往文獻中NDP基因拷貝數變異在不同性別中的臨床表現。
對于本家系臨床診斷是ND還是FEVR,我們想追蹤家系中其他成員是否存在有聽力和智力異常,再進一步確診,但先證者母親不愿做臨床評估。本家系確診病例中,臨床表現不能明確是哪一種疾病,我們考慮是否可從第2號外顯子的缺失,影響蛋白質功能方面分析其臨床表型。
NDP基因跨越28 kb基因組,包含3個外顯子,編碼部分跨越第2號外顯子的后半部分和第3號外顯子的第一部分,開放閱讀框的前58個殘基位于第2號外顯子,而開放閱讀框的密碼子59~133和3'非翻譯區位于第3號外顯子[12]。Norrie的胱氨酸結結構域從32~133密碼子,被認為在神經相互作用中發揮重要作用[13]。在密碼子39、65、69、96、126、128處的半胱氨酸殘基被發現負責半胱氨酸結的形成。密碼子39和96、65和126與密碼子69和128之間的3個二硫橋參與了Norrin的三級結構[1]。
Wu等[1]分析了5例ND患者和4例FEVR患者的NDP基因變異,發現ND患者的變異涉及Norrie蛋白的半胱氨酸殘基,而Norrie蛋白的非半胱氨酸殘基突變顯示血管和視網膜發育異常,更符合X連鎖遺傳的FEVR。破壞半胱氨酸結基序的突變與嚴重視網膜發育不良相對應,而非半胱氨酸突變患者有不同程度的無血管周圍視網膜、視網膜外血管系統和視網膜下滲出物。部分NDP基因研究表明,潛在連續缺失較基因內變異(小變異或第2號外顯子缺失)的患者更有可能出現認知障礙和行為表型[6],基因內微結構的缺失更容易引起神經癥狀[14]。
本家系患者的NDP基因第2號外顯子缺失,缺失部位涉及半胱氨酸結構,為ND表現,且家系中應有聽力和神經癥狀表現者,但2例男性患兒表現為嚴重的視網膜脫離和出生后盲,母親無視力異常表現,僅在FFA檢查中發現周邊視網膜的無血管區。既往文獻對于第2號外顯子缺失的報道較少,檢索到的文獻也是視力喪失患者的基因檢測和臨床表現,無家系的臨床表現追蹤[4, 10]。在HGMD中收錄有1例NDP基因第2號外顯子缺失的報道[15],此文獻是對于患者聽力的20年追蹤觀察,患者出生時視力喪失,在青春期才出現感覺性聽力損失。
對于NDP基因突變家系中臨床表現不同的研究,Liu等[16]曾報道1例NDP基因變異所致男性ND患兒。患兒2歲后出現學習和與他人交流方面的困難,同時出現指壓眼征和自傷行為;4歲時因雙側白內障及視力差就診,B型超聲檢查顯示雙眼視網膜脫離。患兒父親有與脊髓灰質炎相關的身體殘疾,眼部檢查未見異常;母親左眼有眼球癆和角膜病,視力無光感;姨媽雙眼眼底檢查正常;基因檢測母親和姨媽NDP基因中存在第2、3號外顯子的純合子缺失,診斷為ND。
因此,本家系也許是ND在家系中雜合和純合的表現型不同,或許就是FEVR,亦或是兩種疾病在表現上的重合。或與部分研究者意見一致,NDP基因突變涉及廣泛的Wnt信號異常,因而FEVR和ND并非總能做出明確診斷[17]。我們將繼續追蹤觀察家系中其他成員的表現,患者聽力、神經以及其他眼外表現,也許多年后才能最終確定是ND或FEVR。
本家系中先證者母親在第三胎懷孕19周后接受了羊膜穿刺基因檢測,未發現胎兒基因突變。妊娠30周時,對胎兒眼部進行了超聲檢查,顯示對稱球狀,透明明亮的晶狀體和玻璃體腔。出生后眼科檢查符合無NDP基因突變表現。本研究結果證實,在遺傳性視網膜疾病家庭中,羊膜穿刺產前診斷是有效阻斷家族遺傳疾病的方式,為懷孕一個健康的孩子提供安全、可靠的幫助。
NDP基因第2號外顯子缺失的臨床表型在一個家系有兩種表現,對于此家系我們還將進行長期隨訪,觀察眼部體征的發展情況,以及眼外的其他體征,為NDP基因突變的基因型和表型的關系研究提供依據。