引用本文: 劉偉偉, 楚瑩瑩, 王惠, 王飛, 楚瑞雪, 孫先桃, 盧躍兵, 余繼鋒. Alstr?m綜合征患者眼部臨床特征和致病基因分析. 中華眼底病雜志, 2023, 39(7): 530-537. doi: 10.3760/cma.j.cn511434-20221018-00552 復制
Alstr?m綜合征(ALMS,OMIM #203800)是一種罕見的常染色體隱性遺傳性纖毛疾病,發病率約為1/1 000 000,由位于染色體2p13的ALMS1基因突變所致,臨床表現極為復雜[1-2]。嬰兒期即可發生因錐桿細胞營養不良(CORD)所致的眼球震顫、畏光,成為多數ALMS患者首發臨床癥狀[1, 3-4]。常見臨床表現還包括感音神經性耳聾、擴張型心肌病、肥胖、胰島素抵抗、2型糖尿病(T2DM)、高脂血癥、肝腎功能損傷、肺纖維化等。因臨床表現多樣化,早期易被誤診,晚期因多器官功能衰竭,且尚無有效治療方法,壽命很少超過50歲[5-7]。目前國內關于ALMS的研究較少,且缺乏詳細的眼部臨床特征描述。本研究對ALMS1基因突變所致ALMS家系3例患者的眼部臨床表現和致病基因進行分析。現將結果報道如下。
1 對象和方法
回顧性臨床研究。本研究通過河南省兒童醫院倫理委員會審核批準(批文號:2022-K-076);嚴格遵循《赫爾辛基宣言》原則;所有受試者及未成年監護人均獲知情并簽署書面知情同意書。
2020年10月至2022年7月于河南省兒童醫院經基因及臨床檢查確診的ALMS患者3例及家系成員5名納入本研究。3例患者來自2個無血緣關系家系。詳細詢問病史、家族史,進行詳細的全身及眼部檢查,并行基因檢測,繪制家系圖(圖1)。受試者均行最佳矯正視力(BCVA)、屈光度、眼前節、眼壓、廣角激光掃描眼底照相、光相干斷層掃描(OCT)、視網膜電圖(ERG)檢查。患者均符合ALMS臨床診斷標準[7]。
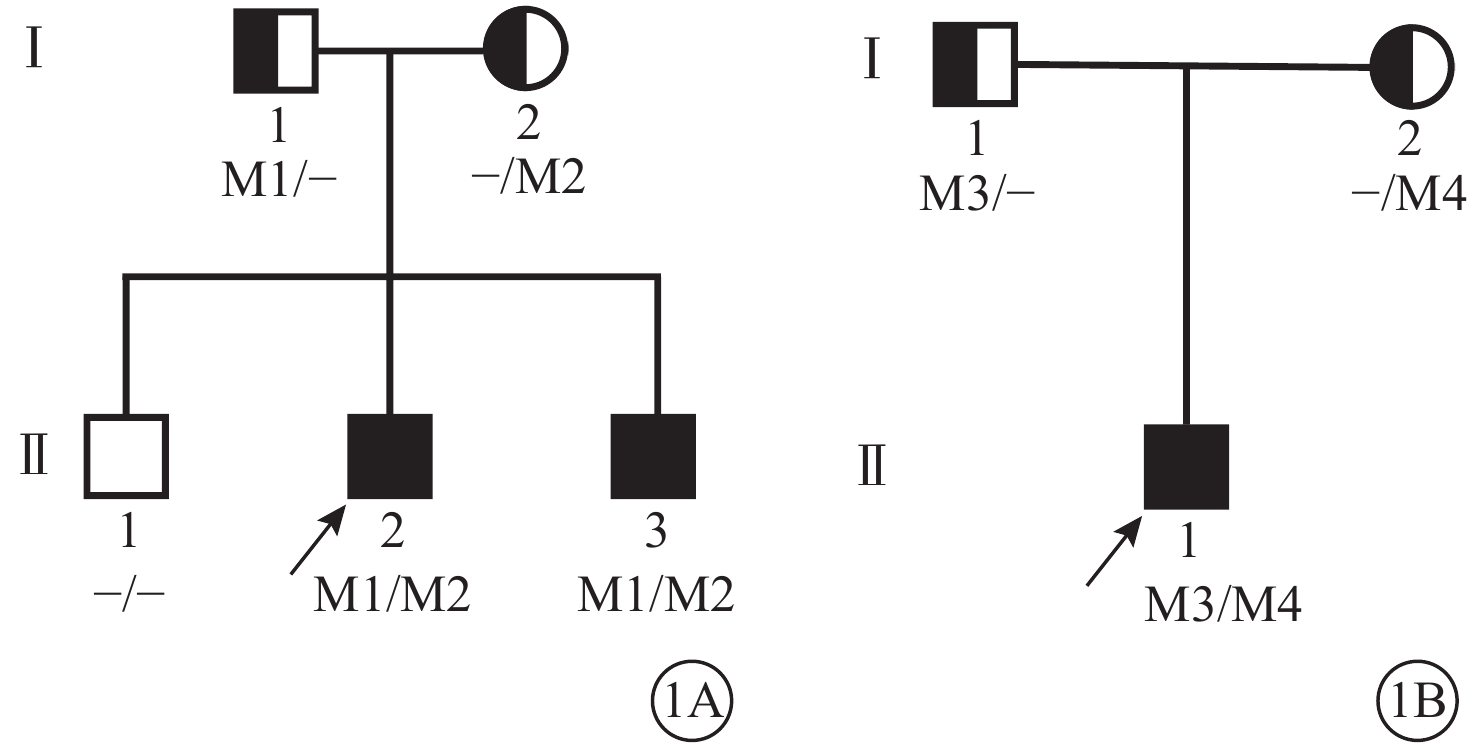
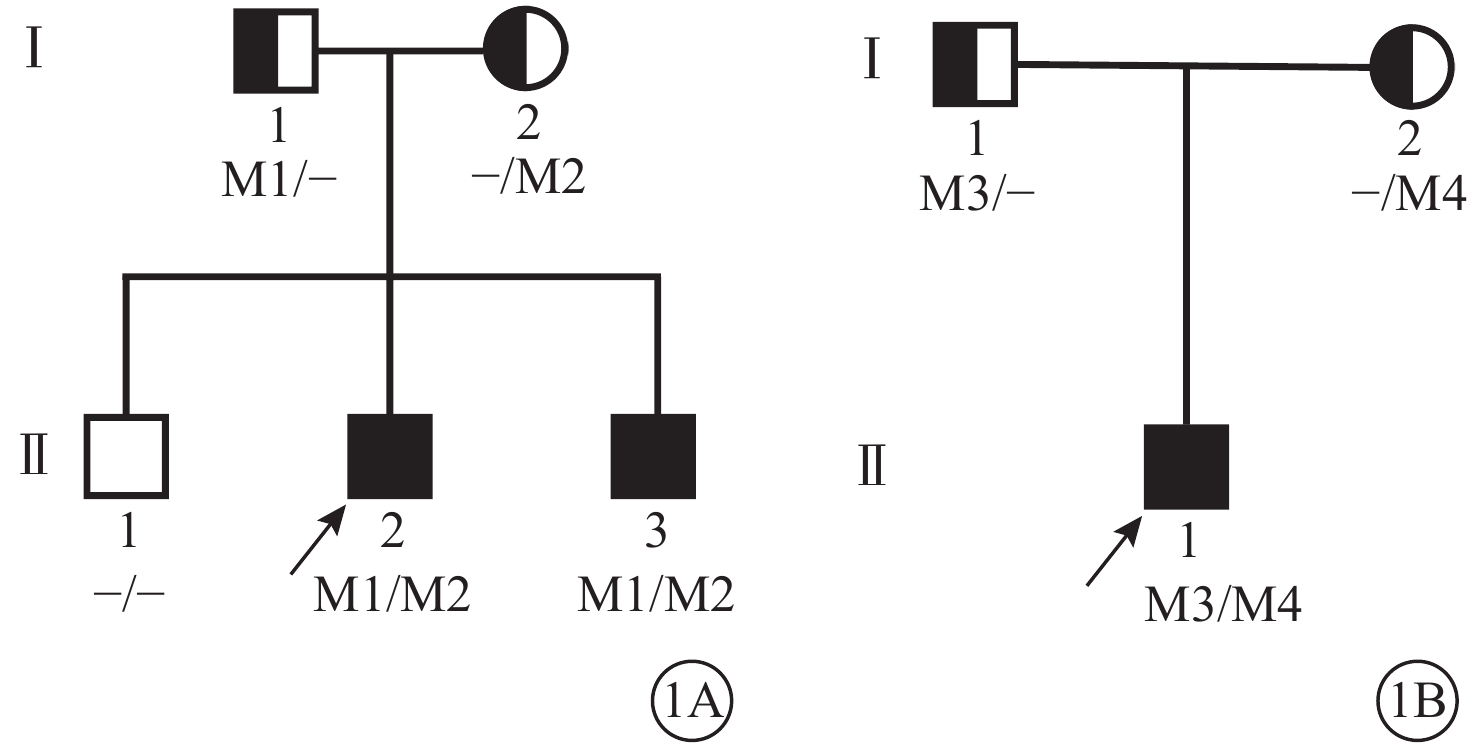 圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。
圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。采集受試者外周靜脈血3 ml,乙二胺四乙酸抗凝。定制安捷倫外顯子組捕獲探針對外顯子區域DNA捕獲并富集,使用高通量測序技術進行致病基因篩查。采用Burrows-Wheeler和UCSC hg19人類參考基因組序列進行比對;使用基因檢測智能操作系統進行變異注釋和解讀,根據人類基因變異數據庫(HGMD,http://www.hgmd.org/)、ClinVar數據庫(https://www.ncbi.nlm.nih.gov/ clinvar/)、OMIM數據庫(https://omim.org/)查找變異位點的收錄情況;應用在線軟件SIFT(http://sift.jcvi.org/)、Polyphen2 (http://genetics.bwh.harvard.edu/pph2/)預測該突變致病性。對檢測出的可疑致病突變,均采用Sanger測序進行驗證。數據解讀規則參考美國醫學遺傳學和基因組學學會基因突變解讀指南[8]。
2 結果
家系1先證者(Ⅱ2),男,14歲。出生3周發現眼球震顫,1歲時畏光,2歲時視力低于同齡兒童;2歲半出現陣發性抽搐,診斷為“癲癇”行藥物治療,3年后停藥。4歲發現外斜視,色覺障礙,外院診斷為“視網膜色素變性、屈光不正”,配鏡治療,隨后視力進行性下降。再后來出現聽力下降,診斷為感音神經性耳聾,現佩戴助聽器。8歲出現多飲、多尿,診斷為T2DM,行降糖治療。因血糖控制不良就診于我院。患兒足月順產,兒童早期肥胖史。全身檢查:身高143.6 cm,體重43.0 kg,體重指數(BMI)20.85 kg/m2;黑棘皮癥,頸部及腋窩明顯;無并指(趾)畸形等。實驗室檢查示肝腎功能異常、高脂血癥、高尿酸血癥、甲狀腺功能異常,性激素各項均正常。葡萄糖耐量試驗(OGTT試驗)提示T2DM,胰島素抵抗。心臟彩色超聲檢查,左心增大,二尖瓣返流,左室收縮功能減低。眼科檢查:角膜映光-40°,眼前節未見明顯異常。BCVA:右眼+6.00 DS/+2.00 DC×90°→無光感,左眼+5.75 DS/+1.75 DC×65°→無光感。眼壓:右眼、左眼分別為14.3、15.6 mm Hg(1 mm Hg=0.133 kPa)。雙眼玻璃體混濁。右眼視盤邊界清楚,顏色尚可;左眼視盤顯示不清。雙眼黃斑結構不清晰,視網膜血管變細、減少,沿血管可見少量點、線狀出血灶,視網膜色素減少,色澤斑駁。OCT檢查,雙眼視網膜變薄,層間結構不清,光感受器細胞層消失,僅見顆粒狀強反射信號,視網膜色素上皮層萎縮變薄,脈絡膜反射增強(圖2)。ERG檢查,雙眼各波形反應呈熄滅型(圖3)。
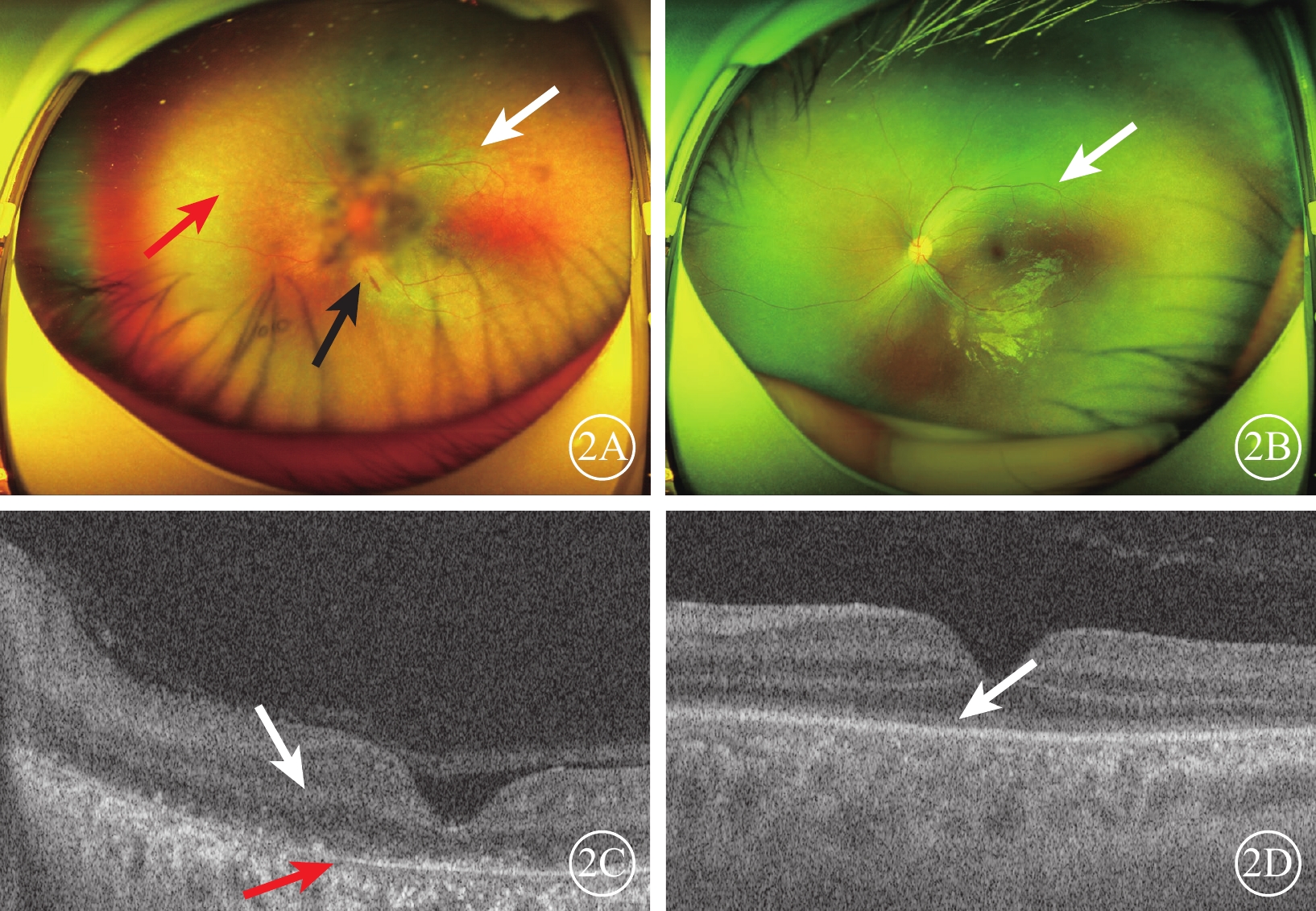
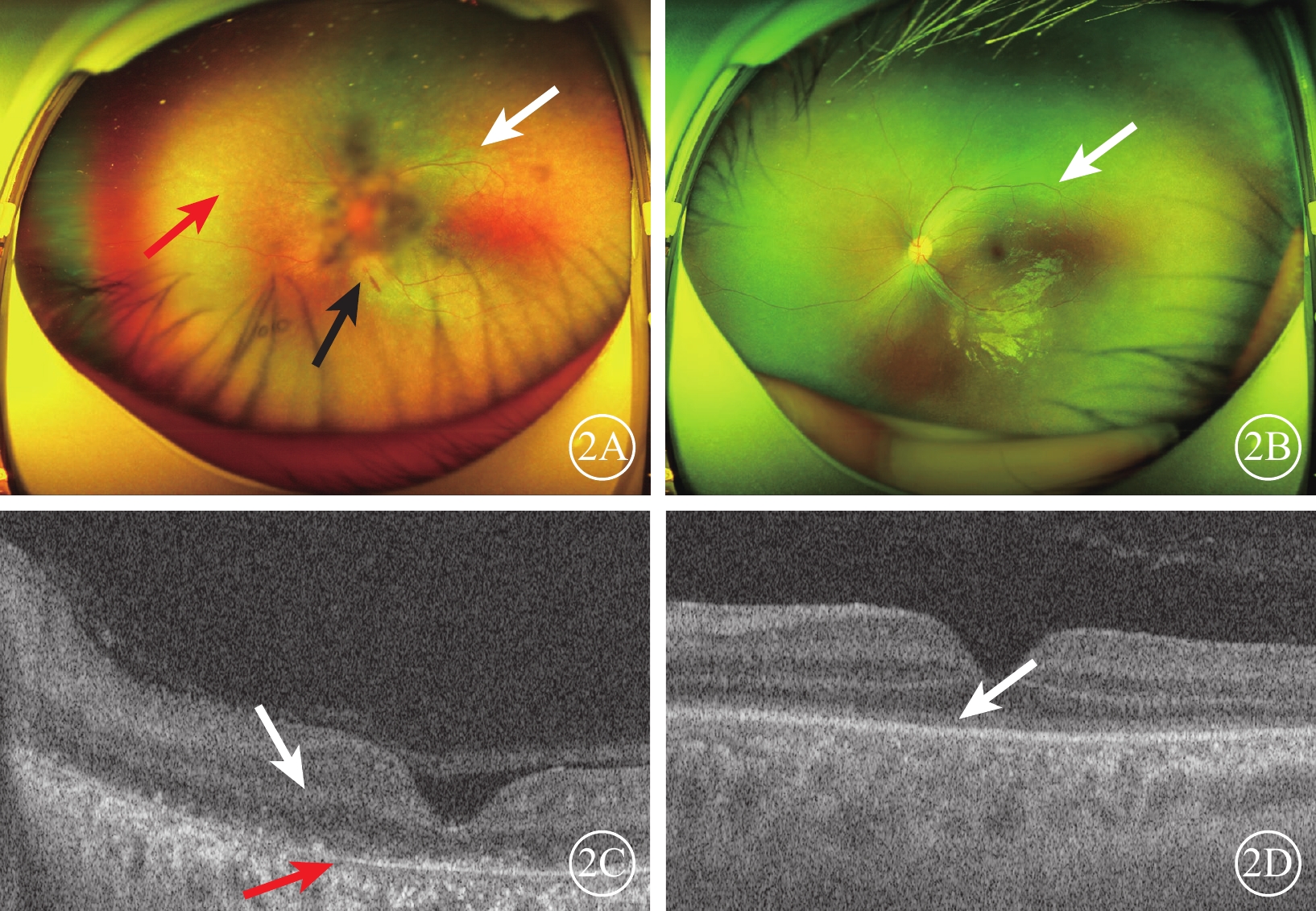 圖2
Alstr?m綜合征家系1先證者(Ⅱ2)及其弟弟(Ⅱ3)彩色眼底、光相干斷層掃描(OCT)像 2A示先證者左眼彩色眼底像,視網膜血管變細、減少(白箭),視網膜色素減少,色澤斑駁(紅箭),沿血管少量點、線狀出血灶(黑箭);2B示先證者弟弟左眼彩色眼底像,視網膜血管變細、減少(白箭);2C示先證者左眼OCT像,黃斑區視網膜層間結構不清(白箭),光感受器細胞層消失,僅見顆粒狀強反射信號,視網膜色素上皮層萎縮變薄(紅箭);2D示先證者弟弟左眼OCT像,光感受器細胞層模糊(白箭)
圖2
Alstr?m綜合征家系1先證者(Ⅱ2)及其弟弟(Ⅱ3)彩色眼底、光相干斷層掃描(OCT)像 2A示先證者左眼彩色眼底像,視網膜血管變細、減少(白箭),視網膜色素減少,色澤斑駁(紅箭),沿血管少量點、線狀出血灶(黑箭);2B示先證者弟弟左眼彩色眼底像,視網膜血管變細、減少(白箭);2C示先證者左眼OCT像,黃斑區視網膜層間結構不清(白箭),光感受器細胞層消失,僅見顆粒狀強反射信號,視網膜色素上皮層萎縮變薄(紅箭);2D示先證者弟弟左眼OCT像,光感受器細胞層模糊(白箭)
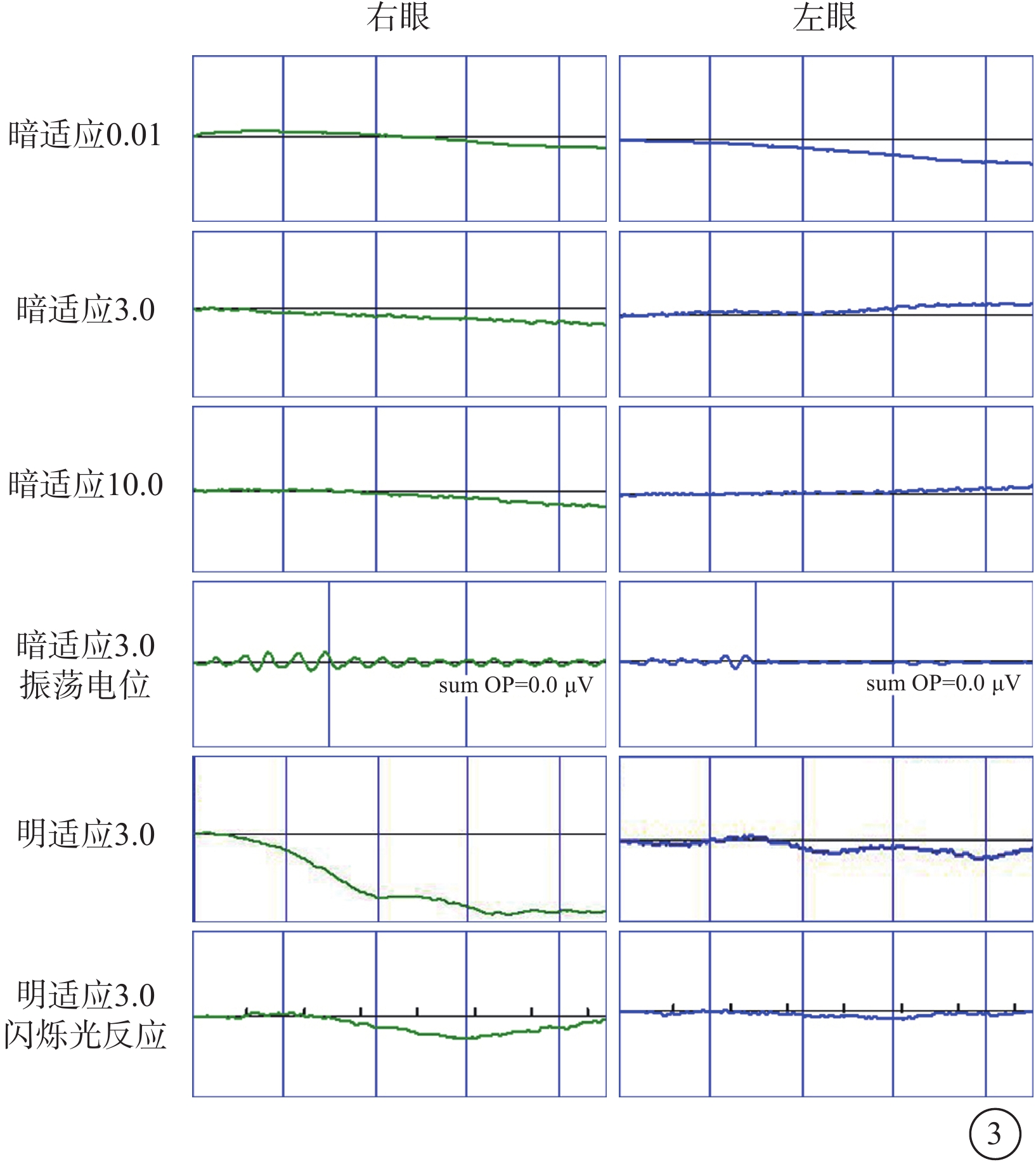
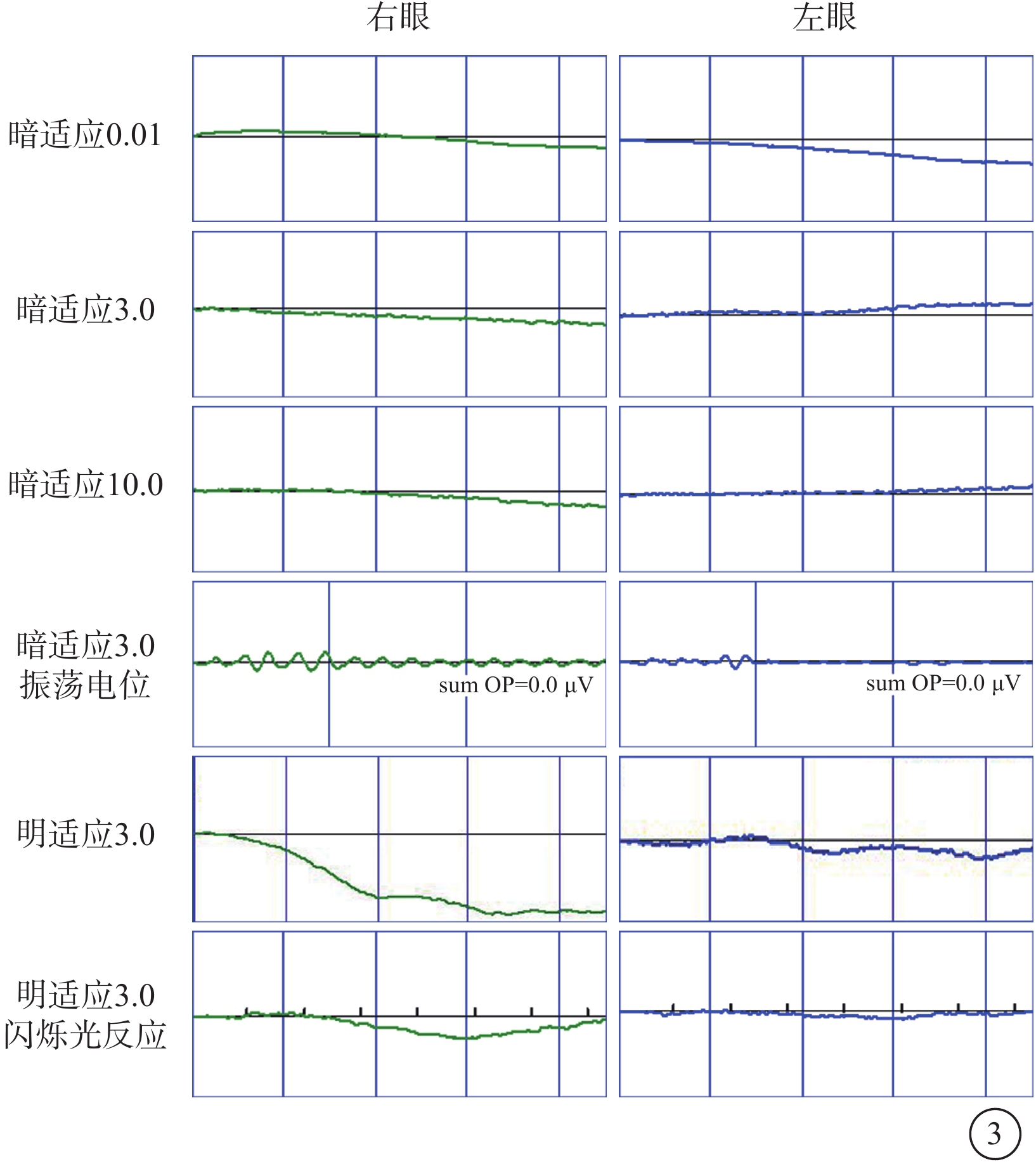 圖3
Alstr?m綜合征家系1先證者(Ⅱ2)視網膜電圖像 各波形呈全熄滅型
圖3
Alstr?m綜合征家系1先證者(Ⅱ2)視網膜電圖像 各波形呈全熄滅型
先證者弟弟(Ⅱ3),7歲。出生1個月時發現眼球震顫,9個月出現畏光,外院診斷為“屈光不正、弱視”,配鏡治療,未行進一步檢查。隨后視力進行性下降,色覺障礙并出現全身脂肪堆積,肥胖。后因聽力下降,被診斷為感音神經性耳聾。全身檢查:身高137.8 cm,體重45.5 kg,BMI 23.96 kg/m2;黑棘皮癥,頸部及腋窩明顯;無并指(趾)畸形等。實驗室檢查示肝功能、甲狀腺功能異常,腎功能、尿酸、血脂及性激素各項指標均正常。OGTT試驗提示胰島素抵抗。心臟彩色超聲、心電圖檢查均未見明顯異常;肝臟彩色超聲檢查,脂肪肝。眼科檢查:角膜映光-15°,可見輕微的水平眼球震顫,眼前節未見明顯異常。BCVA:右眼+2.75 DS/+2.50 DC×95°→0.04,左眼+4.00 DS/+2.50 DC×95°→0.02。雙眼眼壓正常。雙眼視盤邊界清楚,顏色尚可,視網膜血管變細、減少,色素分布大致正常。OCT檢查,雙眼視網膜變薄,以外層變薄明顯,光感受器細胞層模糊,反射減弱(圖2)。ERG檢查,雙眼各波形反應呈熄滅型。先證者父親(Ⅰ1)、母親(Ⅰ2)、兄長(Ⅱ1)眼部及全身檢查均未見明顯異常。
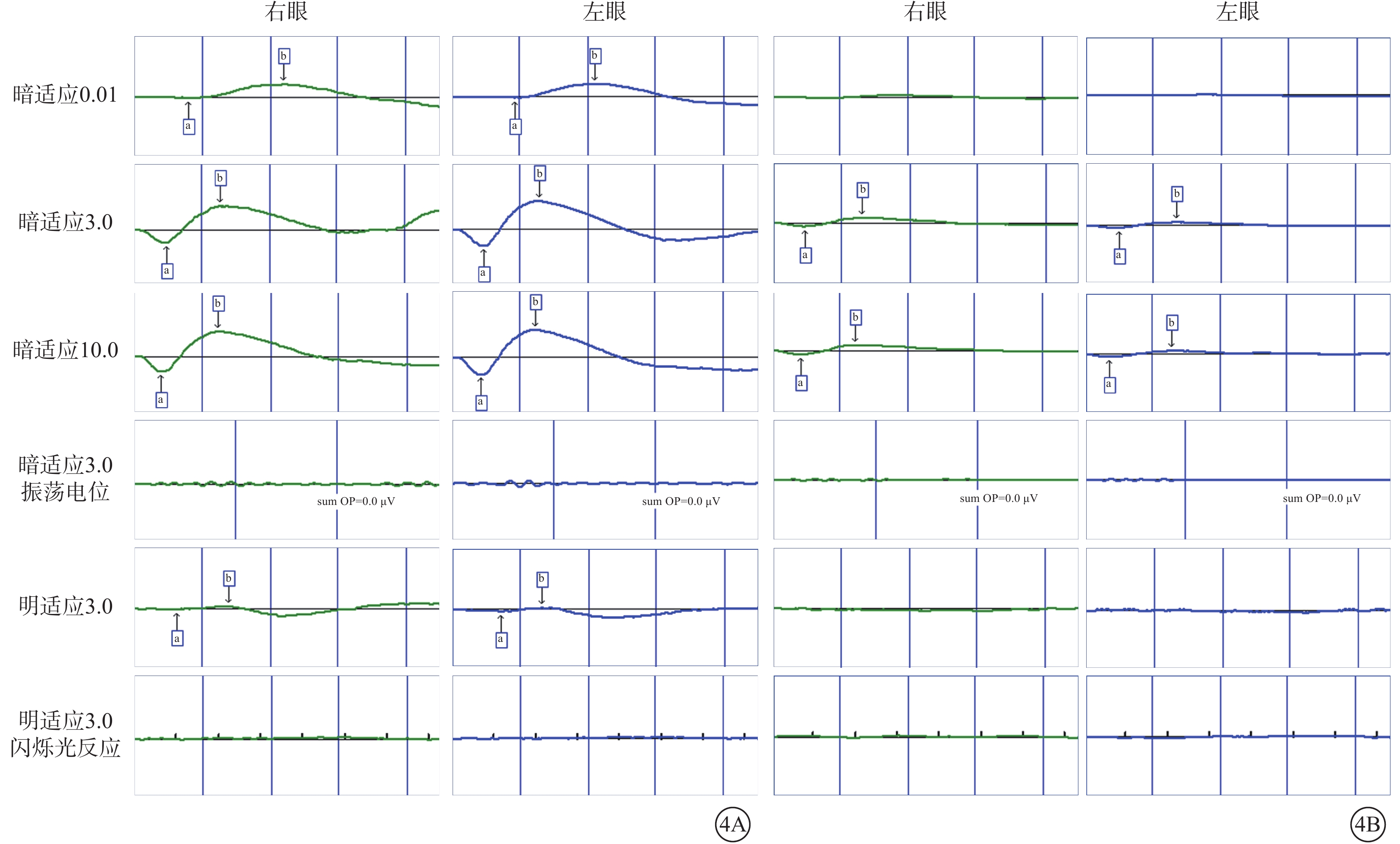
家系2先證者(Ⅱ1),男,23個月。父母非近親結婚,足月順產,出生4個月發現眼球震顫,1歲半出現畏光,偶有失神發作。眼前節及眼底檢查未見明顯異常。ERG檢查,雙眼暗適應0.01:b波輕度降低;暗適應3.0:a、b波輕度降低;暗適應振蕩電位:呈熄滅型;明適應3.0:a、b波重度降低;30 Hz閃爍光反應:呈熄滅型(圖4)。未行特殊治療。4歲時隨訪,BCVA:右眼+4.50 DS/+2.00 DC×90°→0.01,左眼+5.00 DS/+1.50 DC×85°→0.05。雙眼眼壓正常。雙眼視盤邊界清楚,顏色尚可;黃斑中心反光欠清晰,視網膜血管變細、減少,色素大致分布正常(圖5)。ERG檢查,雙眼除暗適應3.0 a、b波重度降低外,其余均呈熄滅型(圖4)。因患兒不能配合未行OCT檢查。患兒聽力、心臟、血糖、血脂、肝腎功能檢查暫無異常。先證者父親(Ⅰ1)、母親(Ⅰ2)眼部及全身檢查均未見明顯異常。
 圖4
家系2先證者(Ⅱ1)視網膜電圖檢查像 4A示患兒23月齡時,雙眼暗適應0.01 b波輕度降低,3.0 a、b波輕度降低,振蕩電位呈熄滅型;明適應3.0 a、b波重度降低,閃爍光反應呈熄滅型。6B示患兒4歲時,雙眼除暗適應3.0 a、b波重度降低外,其余均呈熄滅型
圖4
家系2先證者(Ⅱ1)視網膜電圖檢查像 4A示患兒23月齡時,雙眼暗適應0.01 b波輕度降低,3.0 a、b波輕度降低,振蕩電位呈熄滅型;明適應3.0 a、b波重度降低,閃爍光反應呈熄滅型。6B示患兒4歲時,雙眼除暗適應3.0 a、b波重度降低外,其余均呈熄滅型
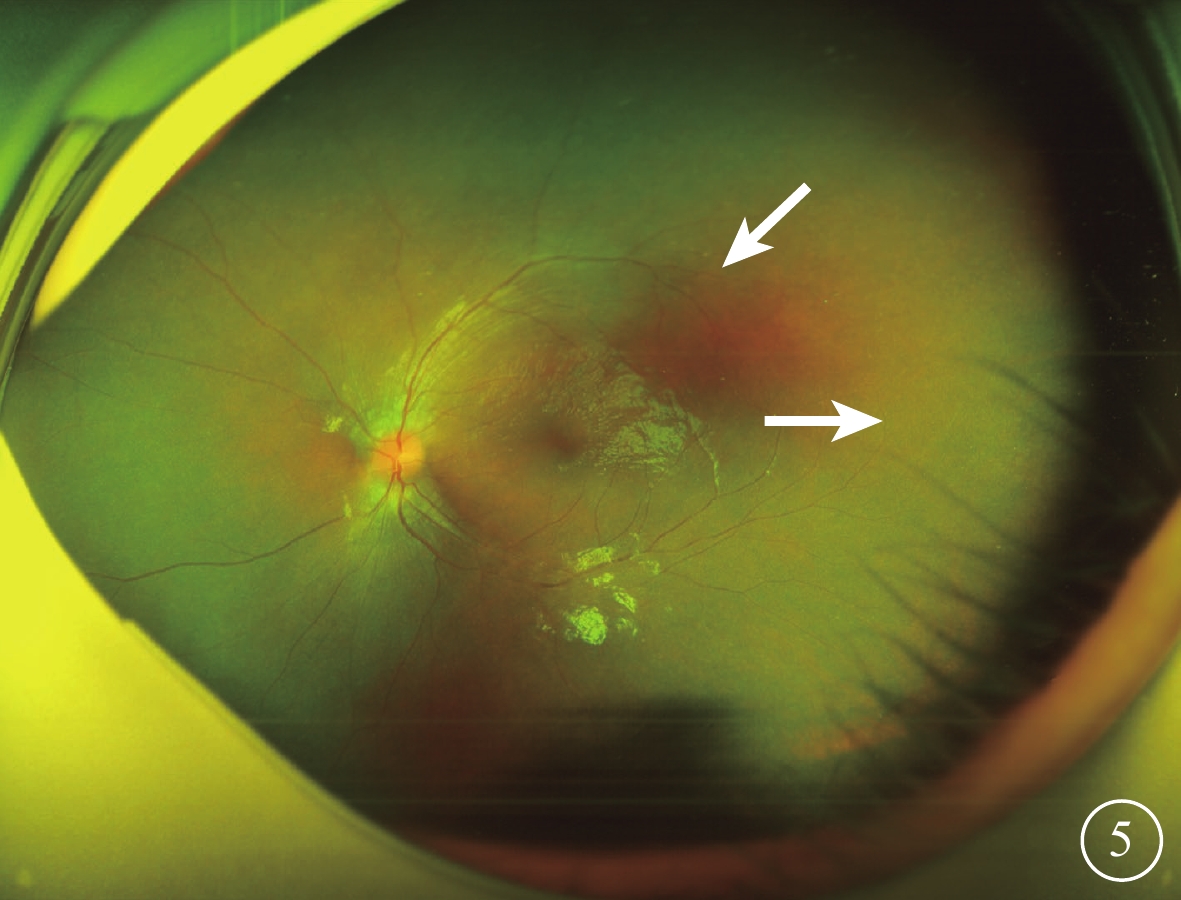
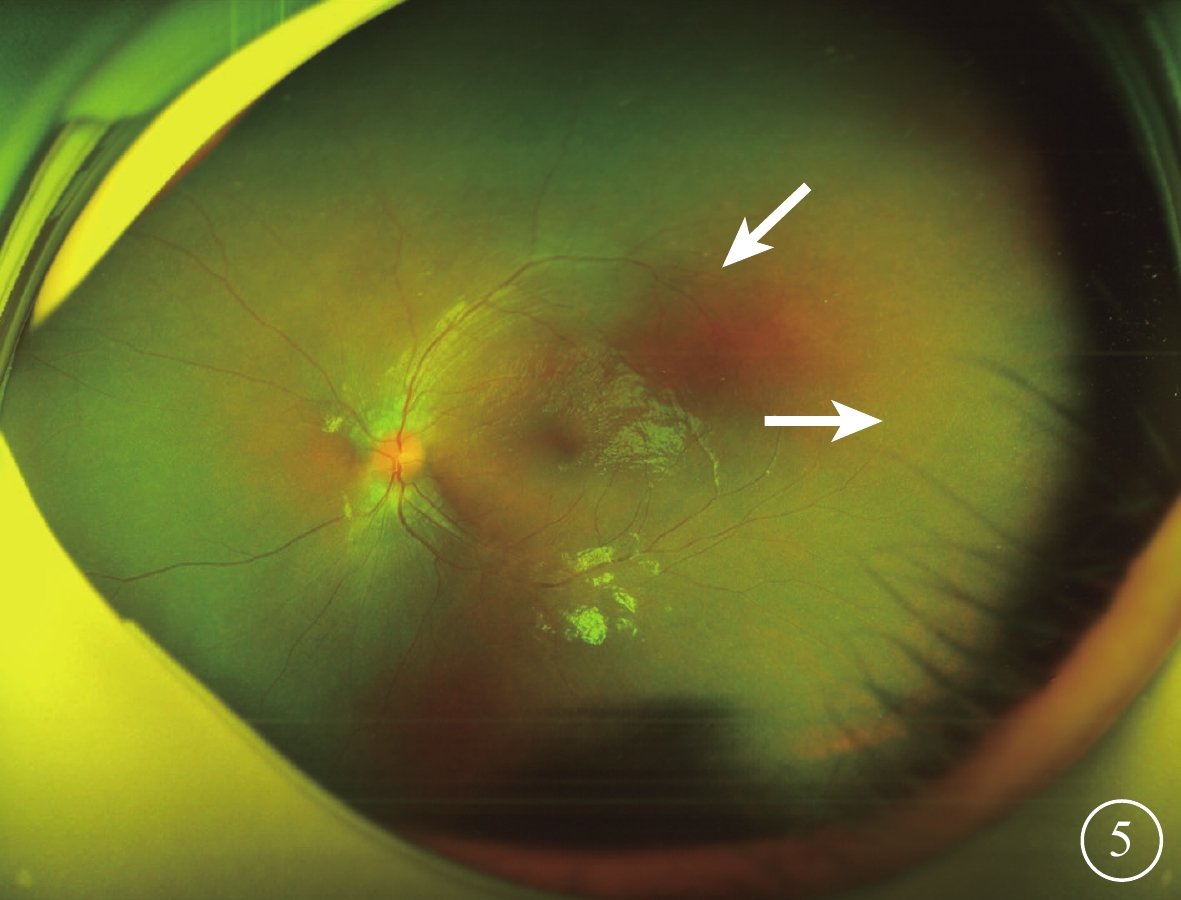 圖5
Alstr?m綜合征家系2先證者(Ⅱ1)左眼彩色眼底像 視網膜血管變細、減少,顳側周邊部血管衰減消失(白箭)
圖5
Alstr?m綜合征家系2先證者(Ⅱ1)左眼彩色眼底像 視網膜血管變細、減少,顳側周邊部血管衰減消失(白箭)
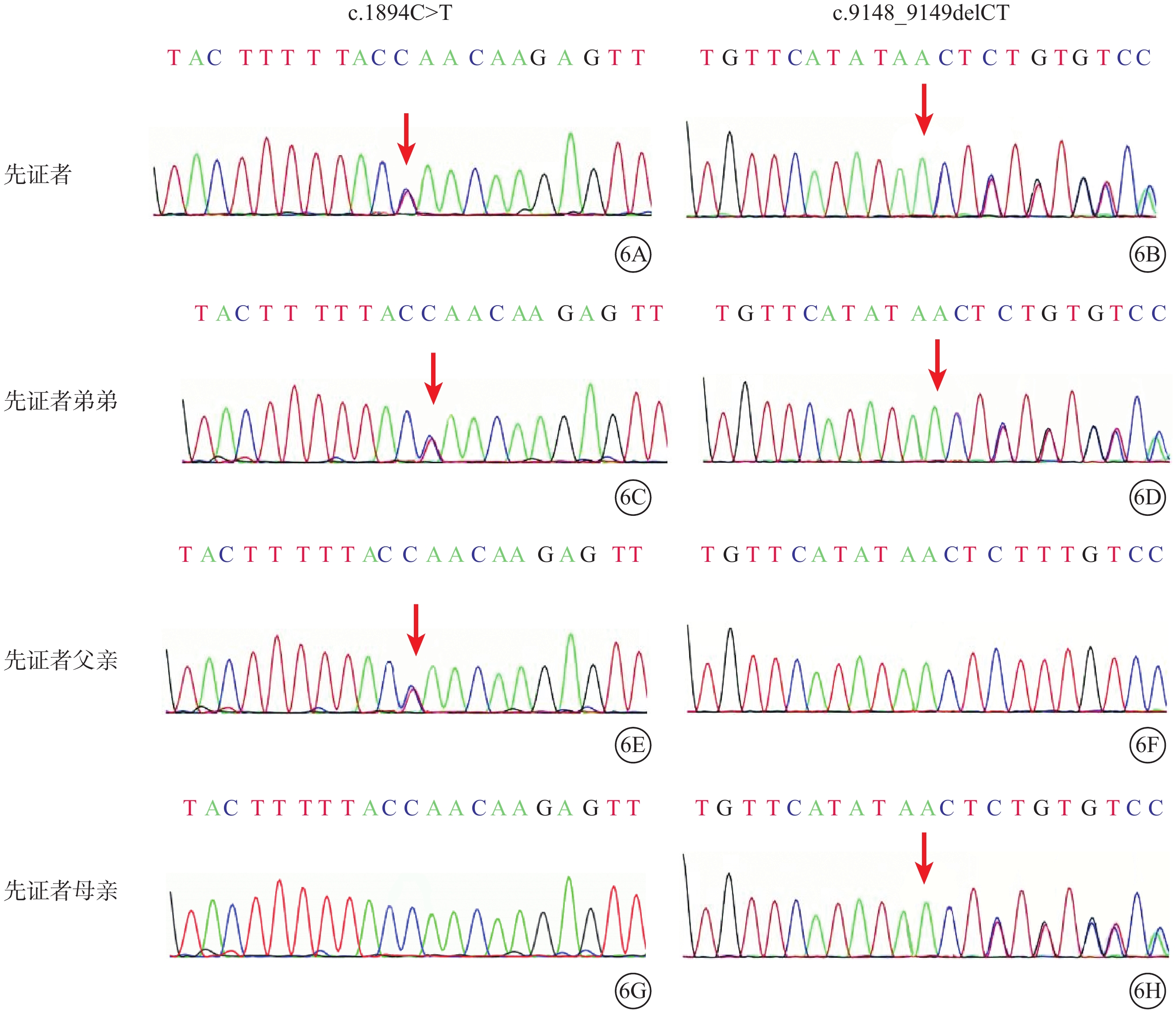
基因檢測結果顯示,家系1先證者(Ⅱ2)及其弟弟(Ⅱ3)ALMS1基因第8、10號外顯子分別存在c.1894C>T/p.Gln632*(M1)、c.9148_9149delCT/p.Leu 3050 Leufs*9(M2)復合雜合突變(圖6)。前者為無義突變;后者為移碼突變,其后第9個氨基酸變為終止密碼子。兩者均使翻譯提前終止,無法生成結構功能正常的蛋白質。M1、M2均已被既往文獻報道[9-10],ClinVar數據庫已收錄。根據ACMG指南,生物學致病等級均被判斷為致病性變異。先證者父親(Ⅰ1)攜帶M1,母親(Ⅰ2)攜帶M2,兄長(Ⅱ1)基因檢測未見異常(圖6)。
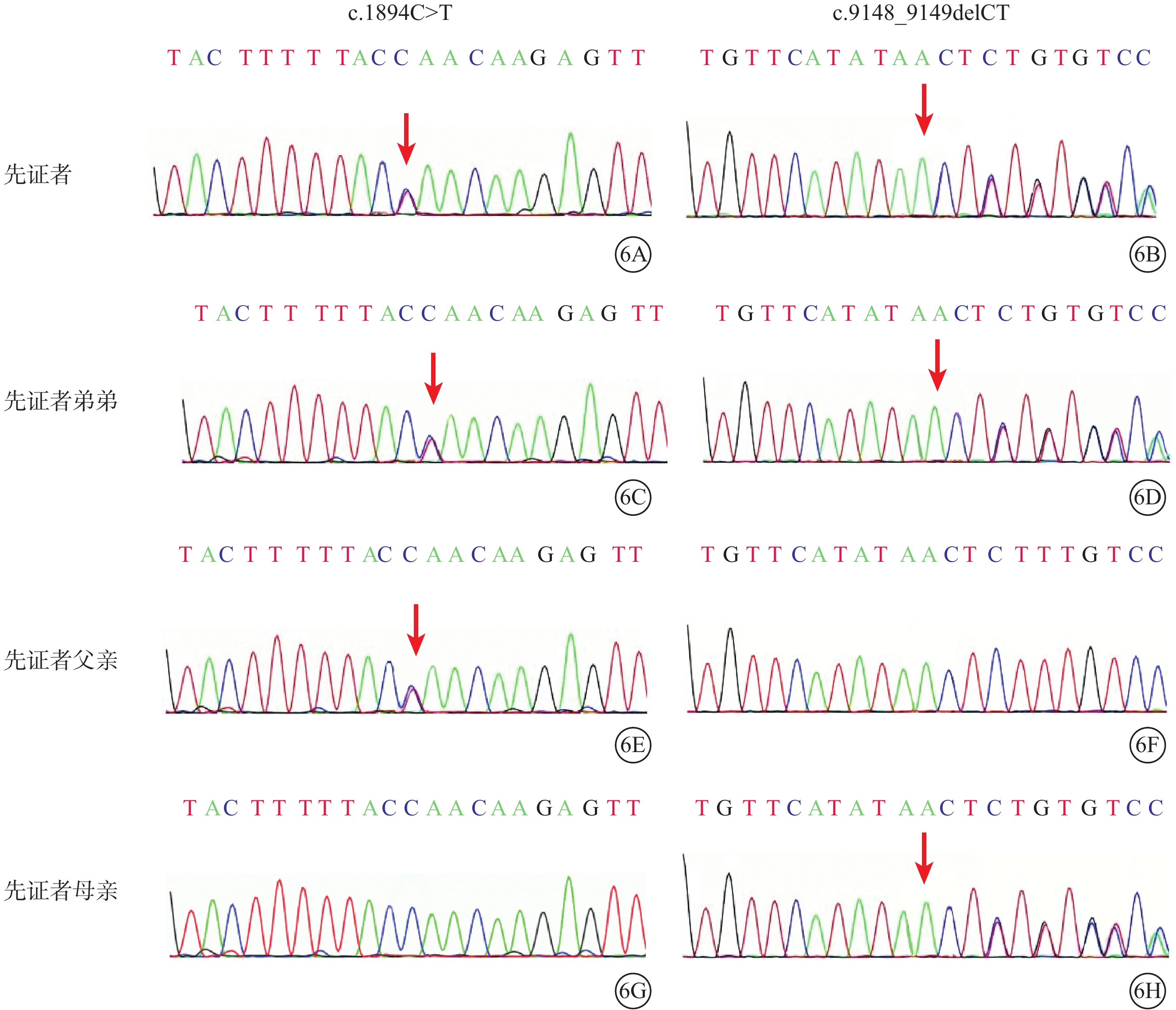 圖6
Alstr?m綜合征家系1基因測序圖 6A、6B和6C、6D分別示先證者及其弟弟,ALMS1基因第8、10號外顯子分別存在c.1894C>T(M1)、c.9148_9149delCT復合雜合突變(紅箭)(M2);6E、6F示先證者父親,攜帶M1突變位點(紅箭);6G、6H示先證者母親,攜帶M2突變位點(紅箭)
圖6
Alstr?m綜合征家系1基因測序圖 6A、6B和6C、6D分別示先證者及其弟弟,ALMS1基因第8、10號外顯子分別存在c.1894C>T(M1)、c.9148_9149delCT復合雜合突變(紅箭)(M2);6E、6F示先證者父親,攜帶M1突變位點(紅箭);6G、6H示先證者母親,攜帶M2突變位點(紅箭)
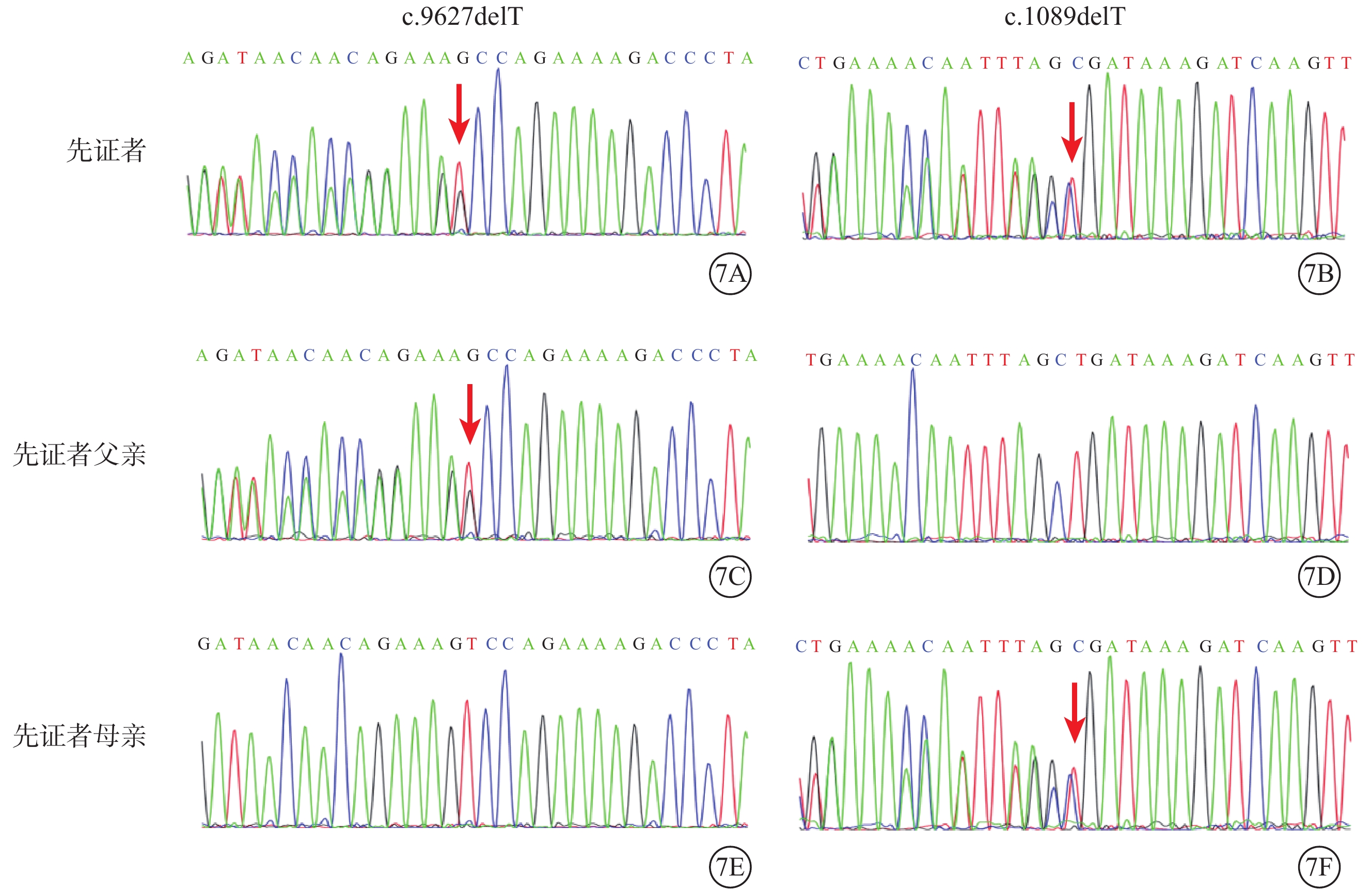
家系2先證者(Ⅱ1)ALMS1基因第11、5號外顯子分別存在c.9627delT/p.Pro3210Glnfs*22(M3)和c.1089delT/p.Asp364Ilefs*13(M4)復合雜合突變(圖7)。兩者均為移碼突變,分別在其后的第22、13個氨基酸變為終止密碼子,使翻譯提前結束,影響蛋白正常功能。M3、M4未見報道,為新發突變位點,正常人群數據庫中未被收錄。根據ACMG指南,生物學致病等級均被判斷為可能致病性變異。先證者父親(Ⅰ1)攜帶M3,母親(Ⅰ2)攜帶M4(圖7)。
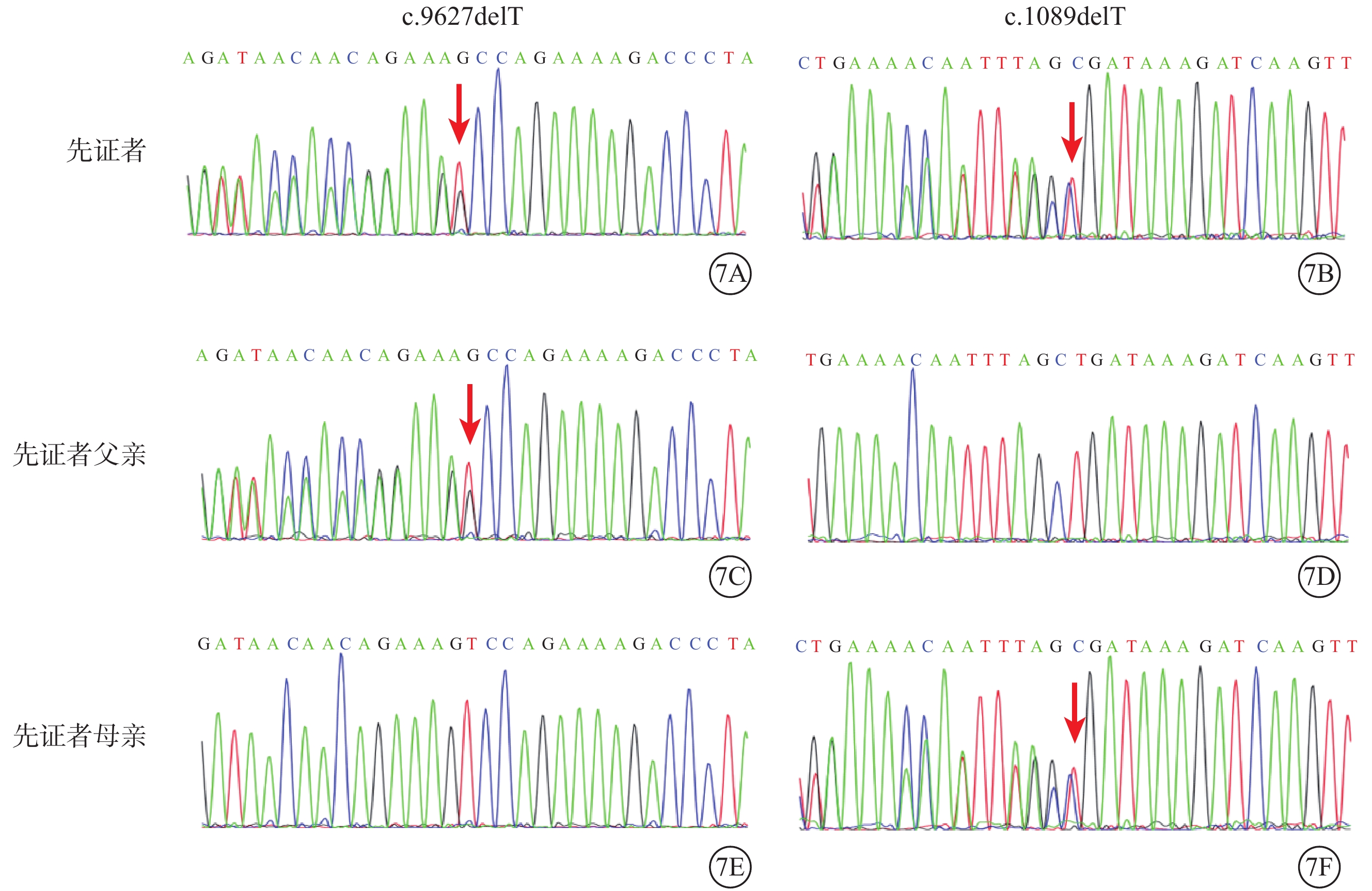 圖7
Alstr?m綜合征家系2基因測序圖 7A、7B示先證者,ALMS1基因第11、5號外顯子分別存在c.9627delT(M3)、c.1089delT(M4)復合雜合突變(紅箭);7C、7D示先證者父親,攜帶M3突變位點(紅箭);7E、7F示先證者母親,攜帶M4突變位點(紅箭)
圖7
Alstr?m綜合征家系2基因測序圖 7A、7B示先證者,ALMS1基因第11、5號外顯子分別存在c.9627delT(M3)、c.1089delT(M4)復合雜合突變(紅箭);7C、7D示先證者父親,攜帶M3突變位點(紅箭);7E、7F示先證者母親,攜帶M4突變位點(紅箭)
3 討論
ALMS又稱視網膜變性-糖尿病-耳聾綜合征,是由ALMS1基因突變引起的罕見全身系統性疾病[2]。臨床表現隨年齡增長發生變化,出生的第一年內可發生視力障礙、嬰兒型擴張性心肌病;兒童期出現肥胖、神經性耳聾、胰島素抵抗、黑棘皮癥;青春期主要表現為T2DM、復發或首發的擴張性心肌病、高脂血癥;成年后身材矮小、肝腎功能損傷、高血壓、男性性功能減退、女性高雄激素血癥、高尿酸血癥、甲狀腺功能異常,疾病晚期出現肺、心臟、肝臟、腎臟等多器官纖維化[7, 11-12]。目前診斷主要依據各階段臨床表現及基因檢測。
ALMS1基因位于2p13.1,由23個外顯子組成,編碼4 169個氨基酸。其廣泛表達于光感受器、聽覺系統、心臟、中樞神經系統、內分泌系統、生殖泌尿系統的纖毛細胞中,在維持細胞內運輸、蛋白質轉運等功能中起重要作用[10, 13-14]。既往研究發現,ALMS多為ALMS1基因發生無義突變和移碼突變,造成蛋白翻譯的提前終止從而失去功能,所以目前多數學者將該病歸為纖毛病[15-17]。ALMS復雜的臨床表現與ALMS1基因的廣泛分布有密切關系。目前已報道近300個ALMS1基因突變位點,熱點突變位于8(51.5%)、16(17.3%)、10(16.0%)號外顯子上,可能與其有較長的編碼序列相關[12-13, 18] 。本研究2個家系中存在4個ALMS1基因突變位點,其中1個無義突變,3個移碼突變。其中M1、M2為已知突變[9-10];M3和M4為新發突變,目前未見報道,增加了ALMS1基因的突變譜。
CORD引起的眼球震顫、畏光、進行性視力下降幾乎發生于所有ALMS患者[13, 19]。患者10歲前可能保存部分有用視力,但視力喪失往往發生在20歲以內[20]。在研究過程中,我們復習了在PubMed檢索到的文中有較為詳細ALMS患者眼部及全身臨床表現的14篇文獻[4, 13, 21-32],總結了文中64例ALSM患者的臨床特征。所有患者中以眼部臨床表現為首發癥狀占75.0%,以心肌病為首發癥狀占23.4%,1歲以內發病占83.7%。所有患者均出現不同程度視力障礙,眼球震顫者92.2%,畏光者90.6%,遠視性屈光不正者91.7%。視力≥0.3者1.9%,0.05~<0.3者32.7%,<0.05者65.4%。后囊下白內障占37.1%。伴隨神經性耳聾占57.1%,心肌病占56.1%,肥胖占84.1%,黑棘皮癥占24.1%,胰島素抵抗/糖尿病占46.7%,高血脂占34.6%,肝功能損傷占30.9%,腎功能損傷占23.2%。本研究中3例患者均在嬰兒期出現眼球震顫及畏光,且為首發臨床表現,并伴隨遠視性屈光不正,BCVA差。家系1中2例患者均出現神經性耳聾、肥胖、黑棘皮癥、胰島素抵抗/糖尿病、肝功能損傷、甲狀腺功能異常,其中先證者還發生了左心擴大伴隨心功能減退、高脂血癥、腎功能損傷、高尿酸血癥等異常。3例患者的臨床表現均符合ALMS的臨床診斷標準[7]。
ALMS患者早期眼底檢查可為正常,但是ERG往往先出現異常。表現為視錐細胞功能不良,隨后視桿受累,最終呈現全熄滅型[27]。隨著疾病進展,眼底也逐漸表現為黃斑中心凹反光不清,視網膜血管變細、減少,色澤斑駁,可伴視神經蒼白。Dotan等[33]總結了22例ALMS者的OCT圖像特征,顯示早期黃斑中心凹變化輕微,隨后光感受器和視網膜色素上皮層進行性丟失,出現視網膜萎縮變薄,以外層顯著,視網膜各層可存在強反射信號,也可伴視網膜皺褶、玻璃體后脫離等。本研究家系2先證者23月齡時眼底檢查正常,但ERG出現了暗適應0.01 b波和3.0 a、b波均輕度降低,明適應3.0 a、b波重度降低。4歲隨訪時,眼底出現了視網膜血管變細衰減,黃斑中心凹反光不清。ERG表現較前加重,除暗適應3.0 a、b波重度降低外,其余均呈熄滅型改變,這再次驗證了ALMS的進行性發展。家系1患先證者弟弟眼底僅表現為視網膜血管的衰減,但OCT已顯示光感受器細胞層模糊,ERG呈熄滅型,視力嚴重受損。所以相比眼底檢查,早期ERG及OCT檢查對于病情的評估可能更為重要。Wang等[32]近期通過對5個家系6例ALMS患者進行分析,也證明了ERG和OCT對于ALMS診斷的重要性。另外,本研究中家系1先證者除典型視網膜血管、色素改變外,還出現沿血管少量點、線狀出血灶,考慮可能與其長期糖尿病且血糖控制不良相關。但在ALMS患者中,并不缺乏T2DM者,眼底出血卻少有報道,或許與ALMS患者的平均壽命有關,值得臨床進一步思考。
ALMS因早期缺乏全身表現,容易被誤診為Leber先天性黑矇、視網膜色素變性、全色盲、孤立性CORD等,這些遺傳性眼底疾病均可表現為早期的眼球震顫、畏光等[4]。Leber先天性黑矇患兒1歲以內即出現不追視,ERG顯示視錐視桿均為熄滅型;視網膜色素變性中夜盲為其特征,早期以視桿細胞損害為主,但ALMS患兒很少出現夜盲,可能是因為當視桿細胞受到損傷時患兒的視力已經非常差,反而感受不到暗視力的變化;全色盲是一種視錐細胞功能障礙性疾病,ERG通常顯示視錐細胞反應呈熄滅型,視桿細胞反應基本正常;ALMS通常比孤立性CORD患者經歷更嚴重且更早的視力損害[4]。當患者出現全身癥狀時,主要與Bardet-Biedl 綜合征(BBS)進行鑒別,BBS也是一種纖毛疾病,除CORD、肥胖、心肌病、腎功能異常、性功能減退、糖耐量異常外,還多出現多指或并指、智力低下等癥狀;另外,BBS患者的視力障礙出現在8歲左右,相對較ALMS更晚[13]。
CORD和嬰兒型擴張型心肌病是ALMS發生最早的臨床表現[11, 13]。我們總結的64例患者中以眼部表現為首發癥狀者占75.0%,這決定了眼科醫師對ALMS診斷的重要性。理想情況下發生眼球震顫的患兒均應接受全面眼科檢查,包括ERG和OCT,以及基因檢測,但因為家長對鎮靜的擔心和較高的基因檢測費用,使得一些遺傳性疾病無法得到早期診治。ALMS雖尚無有效治療方法,但早期診斷可進行心肌病的早期預防,學習盲文,訓練行動技能,通過日常飲食及運動改善肥胖,延緩T2DM發病時間等,可提高患者的生活質量。更多的治療ALMS的方法正在研究中[34]。
國內現有ALMS文獻中,均未詳細描述眼部表現。因該病臨床極為罕見,本研究僅納入我院確診的2個家系中的3例ALMS患者,并進行文獻復習,總結該病的眼部臨床特征,為臨床工作提供參考。目前仍缺乏長期隨訪的臨床資料,期待更多大樣本臨床研究對ALMS進行深入探討。
Alstr?m綜合征(ALMS,OMIM #203800)是一種罕見的常染色體隱性遺傳性纖毛疾病,發病率約為1/1 000 000,由位于染色體2p13的ALMS1基因突變所致,臨床表現極為復雜[1-2]。嬰兒期即可發生因錐桿細胞營養不良(CORD)所致的眼球震顫、畏光,成為多數ALMS患者首發臨床癥狀[1, 3-4]。常見臨床表現還包括感音神經性耳聾、擴張型心肌病、肥胖、胰島素抵抗、2型糖尿病(T2DM)、高脂血癥、肝腎功能損傷、肺纖維化等。因臨床表現多樣化,早期易被誤診,晚期因多器官功能衰竭,且尚無有效治療方法,壽命很少超過50歲[5-7]。目前國內關于ALMS的研究較少,且缺乏詳細的眼部臨床特征描述。本研究對ALMS1基因突變所致ALMS家系3例患者的眼部臨床表現和致病基因進行分析。現將結果報道如下。
1 對象和方法
回顧性臨床研究。本研究通過河南省兒童醫院倫理委員會審核批準(批文號:2022-K-076);嚴格遵循《赫爾辛基宣言》原則;所有受試者及未成年監護人均獲知情并簽署書面知情同意書。
2020年10月至2022年7月于河南省兒童醫院經基因及臨床檢查確診的ALMS患者3例及家系成員5名納入本研究。3例患者來自2個無血緣關系家系。詳細詢問病史、家族史,進行詳細的全身及眼部檢查,并行基因檢測,繪制家系圖(圖1)。受試者均行最佳矯正視力(BCVA)、屈光度、眼前節、眼壓、廣角激光掃描眼底照相、光相干斷層掃描(OCT)、視網膜電圖(ERG)檢查。患者均符合ALMS臨床診斷標準[7]。
圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。采集受試者外周靜脈血3 ml,乙二胺四乙酸抗凝。定制安捷倫外顯子組捕獲探針對外顯子區域DNA捕獲并富集,使用高通量測序技術進行致病基因篩查。采用Burrows-Wheeler和UCSC hg19人類參考基因組序列進行比對;使用基因檢測智能操作系統進行變異注釋和解讀,根據人類基因變異數據庫(HGMD,http://www.hgmd.org/)、ClinVar數據庫(https://www.ncbi.nlm.nih.gov/ clinvar/)、OMIM數據庫(https://omim.org/)查找變異位點的收錄情況;應用在線軟件SIFT(http://sift.jcvi.org/)、Polyphen2 (http://genetics.bwh.harvard.edu/pph2/)預測該突變致病性。對檢測出的可疑致病突變,均采用Sanger測序進行驗證。數據解讀規則參考美國醫學遺傳學和基因組學學會基因突變解讀指南[8]。
2 結果
家系1先證者(Ⅱ2),男,14歲。出生3周發現眼球震顫,1歲時畏光,2歲時視力低于同齡兒童;2歲半出現陣發性抽搐,診斷為“癲癇”行藥物治療,3年后停藥。4歲發現外斜視,色覺障礙,外院診斷為“視網膜色素變性、屈光不正”,配鏡治療,隨后視力進行性下降。再后來出現聽力下降,診斷為感音神經性耳聾,現佩戴助聽器。8歲出現多飲、多尿,診斷為T2DM,行降糖治療。因血糖控制不良就診于我院。患兒足月順產,兒童早期肥胖史。全身檢查:身高143.6 cm,體重43.0 kg,體重指數(BMI)20.85 kg/m2;黑棘皮癥,頸部及腋窩明顯;無并指(趾)畸形等。實驗室檢查示肝腎功能異常、高脂血癥、高尿酸血癥、甲狀腺功能異常,性激素各項均正常。葡萄糖耐量試驗(OGTT試驗)提示T2DM,胰島素抵抗。心臟彩色超聲檢查,左心增大,二尖瓣返流,左室收縮功能減低。眼科檢查:角膜映光-40°,眼前節未見明顯異常。BCVA:右眼+6.00 DS/+2.00 DC×90°→無光感,左眼+5.75 DS/+1.75 DC×65°→無光感。眼壓:右眼、左眼分別為14.3、15.6 mm Hg(1 mm Hg=0.133 kPa)。雙眼玻璃體混濁。右眼視盤邊界清楚,顏色尚可;左眼視盤顯示不清。雙眼黃斑結構不清晰,視網膜血管變細、減少,沿血管可見少量點、線狀出血灶,視網膜色素減少,色澤斑駁。OCT檢查,雙眼視網膜變薄,層間結構不清,光感受器細胞層消失,僅見顆粒狀強反射信號,視網膜色素上皮層萎縮變薄,脈絡膜反射增強(圖2)。ERG檢查,雙眼各波形反應呈熄滅型(圖3)。
圖2
Alstr?m綜合征家系1先證者(Ⅱ2)及其弟弟(Ⅱ3)彩色眼底、光相干斷層掃描(OCT)像 2A示先證者左眼彩色眼底像,視網膜血管變細、減少(白箭),視網膜色素減少,色澤斑駁(紅箭),沿血管少量點、線狀出血灶(黑箭);2B示先證者弟弟左眼彩色眼底像,視網膜血管變細、減少(白箭);2C示先證者左眼OCT像,黃斑區視網膜層間結構不清(白箭),光感受器細胞層消失,僅見顆粒狀強反射信號,視網膜色素上皮層萎縮變薄(紅箭);2D示先證者弟弟左眼OCT像,光感受器細胞層模糊(白箭)
圖3
Alstr?m綜合征家系1先證者(Ⅱ2)視網膜電圖像 各波形呈全熄滅型
先證者弟弟(Ⅱ3),7歲。出生1個月時發現眼球震顫,9個月出現畏光,外院診斷為“屈光不正、弱視”,配鏡治療,未行進一步檢查。隨后視力進行性下降,色覺障礙并出現全身脂肪堆積,肥胖。后因聽力下降,被診斷為感音神經性耳聾。全身檢查:身高137.8 cm,體重45.5 kg,BMI 23.96 kg/m2;黑棘皮癥,頸部及腋窩明顯;無并指(趾)畸形等。實驗室檢查示肝功能、甲狀腺功能異常,腎功能、尿酸、血脂及性激素各項指標均正常。OGTT試驗提示胰島素抵抗。心臟彩色超聲、心電圖檢查均未見明顯異常;肝臟彩色超聲檢查,脂肪肝。眼科檢查:角膜映光-15°,可見輕微的水平眼球震顫,眼前節未見明顯異常。BCVA:右眼+2.75 DS/+2.50 DC×95°→0.04,左眼+4.00 DS/+2.50 DC×95°→0.02。雙眼眼壓正常。雙眼視盤邊界清楚,顏色尚可,視網膜血管變細、減少,色素分布大致正常。OCT檢查,雙眼視網膜變薄,以外層變薄明顯,光感受器細胞層模糊,反射減弱(圖2)。ERG檢查,雙眼各波形反應呈熄滅型。先證者父親(Ⅰ1)、母親(Ⅰ2)、兄長(Ⅱ1)眼部及全身檢查均未見明顯異常。
家系2先證者(Ⅱ1),男,23個月。父母非近親結婚,足月順產,出生4個月發現眼球震顫,1歲半出現畏光,偶有失神發作。眼前節及眼底檢查未見明顯異常。ERG檢查,雙眼暗適應0.01:b波輕度降低;暗適應3.0:a、b波輕度降低;暗適應振蕩電位:呈熄滅型;明適應3.0:a、b波重度降低;30 Hz閃爍光反應:呈熄滅型(圖4)。未行特殊治療。4歲時隨訪,BCVA:右眼+4.50 DS/+2.00 DC×90°→0.01,左眼+5.00 DS/+1.50 DC×85°→0.05。雙眼眼壓正常。雙眼視盤邊界清楚,顏色尚可;黃斑中心反光欠清晰,視網膜血管變細、減少,色素大致分布正常(圖5)。ERG檢查,雙眼除暗適應3.0 a、b波重度降低外,其余均呈熄滅型(圖4)。因患兒不能配合未行OCT檢查。患兒聽力、心臟、血糖、血脂、肝腎功能檢查暫無異常。先證者父親(Ⅰ1)、母親(Ⅰ2)眼部及全身檢查均未見明顯異常。
圖4
家系2先證者(Ⅱ1)視網膜電圖檢查像 4A示患兒23月齡時,雙眼暗適應0.01 b波輕度降低,3.0 a、b波輕度降低,振蕩電位呈熄滅型;明適應3.0 a、b波重度降低,閃爍光反應呈熄滅型。6B示患兒4歲時,雙眼除暗適應3.0 a、b波重度降低外,其余均呈熄滅型
圖5
Alstr?m綜合征家系2先證者(Ⅱ1)左眼彩色眼底像 視網膜血管變細、減少,顳側周邊部血管衰減消失(白箭)
基因檢測結果顯示,家系1先證者(Ⅱ2)及其弟弟(Ⅱ3)ALMS1基因第8、10號外顯子分別存在c.1894C>T/p.Gln632*(M1)、c.9148_9149delCT/p.Leu 3050 Leufs*9(M2)復合雜合突變(圖6)。前者為無義突變;后者為移碼突變,其后第9個氨基酸變為終止密碼子。兩者均使翻譯提前終止,無法生成結構功能正常的蛋白質。M1、M2均已被既往文獻報道[9-10],ClinVar數據庫已收錄。根據ACMG指南,生物學致病等級均被判斷為致病性變異。先證者父親(Ⅰ1)攜帶M1,母親(Ⅰ2)攜帶M2,兄長(Ⅱ1)基因檢測未見異常(圖6)。
圖6
Alstr?m綜合征家系1基因測序圖 6A、6B和6C、6D分別示先證者及其弟弟,ALMS1基因第8、10號外顯子分別存在c.1894C>T(M1)、c.9148_9149delCT復合雜合突變(紅箭)(M2);6E、6F示先證者父親,攜帶M1突變位點(紅箭);6G、6H示先證者母親,攜帶M2突變位點(紅箭)
家系2先證者(Ⅱ1)ALMS1基因第11、5號外顯子分別存在c.9627delT/p.Pro3210Glnfs*22(M3)和c.1089delT/p.Asp364Ilefs*13(M4)復合雜合突變(圖7)。兩者均為移碼突變,分別在其后的第22、13個氨基酸變為終止密碼子,使翻譯提前結束,影響蛋白正常功能。M3、M4未見報道,為新發突變位點,正常人群數據庫中未被收錄。根據ACMG指南,生物學致病等級均被判斷為可能致病性變異。先證者父親(Ⅰ1)攜帶M3,母親(Ⅰ2)攜帶M4(圖7)。
圖7
Alstr?m綜合征家系2基因測序圖 7A、7B示先證者,ALMS1基因第11、5號外顯子分別存在c.9627delT(M3)、c.1089delT(M4)復合雜合突變(紅箭);7C、7D示先證者父親,攜帶M3突變位點(紅箭);7E、7F示先證者母親,攜帶M4突變位點(紅箭)
3 討論
ALMS又稱視網膜變性-糖尿病-耳聾綜合征,是由ALMS1基因突變引起的罕見全身系統性疾病[2]。臨床表現隨年齡增長發生變化,出生的第一年內可發生視力障礙、嬰兒型擴張性心肌病;兒童期出現肥胖、神經性耳聾、胰島素抵抗、黑棘皮癥;青春期主要表現為T2DM、復發或首發的擴張性心肌病、高脂血癥;成年后身材矮小、肝腎功能損傷、高血壓、男性性功能減退、女性高雄激素血癥、高尿酸血癥、甲狀腺功能異常,疾病晚期出現肺、心臟、肝臟、腎臟等多器官纖維化[7, 11-12]。目前診斷主要依據各階段臨床表現及基因檢測。
ALMS1基因位于2p13.1,由23個外顯子組成,編碼4 169個氨基酸。其廣泛表達于光感受器、聽覺系統、心臟、中樞神經系統、內分泌系統、生殖泌尿系統的纖毛細胞中,在維持細胞內運輸、蛋白質轉運等功能中起重要作用[10, 13-14]。既往研究發現,ALMS多為ALMS1基因發生無義突變和移碼突變,造成蛋白翻譯的提前終止從而失去功能,所以目前多數學者將該病歸為纖毛病[15-17]。ALMS復雜的臨床表現與ALMS1基因的廣泛分布有密切關系。目前已報道近300個ALMS1基因突變位點,熱點突變位于8(51.5%)、16(17.3%)、10(16.0%)號外顯子上,可能與其有較長的編碼序列相關[12-13, 18] 。本研究2個家系中存在4個ALMS1基因突變位點,其中1個無義突變,3個移碼突變。其中M1、M2為已知突變[9-10];M3和M4為新發突變,目前未見報道,增加了ALMS1基因的突變譜。
CORD引起的眼球震顫、畏光、進行性視力下降幾乎發生于所有ALMS患者[13, 19]。患者10歲前可能保存部分有用視力,但視力喪失往往發生在20歲以內[20]。在研究過程中,我們復習了在PubMed檢索到的文中有較為詳細ALMS患者眼部及全身臨床表現的14篇文獻[4, 13, 21-32],總結了文中64例ALSM患者的臨床特征。所有患者中以眼部臨床表現為首發癥狀占75.0%,以心肌病為首發癥狀占23.4%,1歲以內發病占83.7%。所有患者均出現不同程度視力障礙,眼球震顫者92.2%,畏光者90.6%,遠視性屈光不正者91.7%。視力≥0.3者1.9%,0.05~<0.3者32.7%,<0.05者65.4%。后囊下白內障占37.1%。伴隨神經性耳聾占57.1%,心肌病占56.1%,肥胖占84.1%,黑棘皮癥占24.1%,胰島素抵抗/糖尿病占46.7%,高血脂占34.6%,肝功能損傷占30.9%,腎功能損傷占23.2%。本研究中3例患者均在嬰兒期出現眼球震顫及畏光,且為首發臨床表現,并伴隨遠視性屈光不正,BCVA差。家系1中2例患者均出現神經性耳聾、肥胖、黑棘皮癥、胰島素抵抗/糖尿病、肝功能損傷、甲狀腺功能異常,其中先證者還發生了左心擴大伴隨心功能減退、高脂血癥、腎功能損傷、高尿酸血癥等異常。3例患者的臨床表現均符合ALMS的臨床診斷標準[7]。
ALMS患者早期眼底檢查可為正常,但是ERG往往先出現異常。表現為視錐細胞功能不良,隨后視桿受累,最終呈現全熄滅型[27]。隨著疾病進展,眼底也逐漸表現為黃斑中心凹反光不清,視網膜血管變細、減少,色澤斑駁,可伴視神經蒼白。Dotan等[33]總結了22例ALMS者的OCT圖像特征,顯示早期黃斑中心凹變化輕微,隨后光感受器和視網膜色素上皮層進行性丟失,出現視網膜萎縮變薄,以外層顯著,視網膜各層可存在強反射信號,也可伴視網膜皺褶、玻璃體后脫離等。本研究家系2先證者23月齡時眼底檢查正常,但ERG出現了暗適應0.01 b波和3.0 a、b波均輕度降低,明適應3.0 a、b波重度降低。4歲隨訪時,眼底出現了視網膜血管變細衰減,黃斑中心凹反光不清。ERG表現較前加重,除暗適應3.0 a、b波重度降低外,其余均呈熄滅型改變,這再次驗證了ALMS的進行性發展。家系1患先證者弟弟眼底僅表現為視網膜血管的衰減,但OCT已顯示光感受器細胞層模糊,ERG呈熄滅型,視力嚴重受損。所以相比眼底檢查,早期ERG及OCT檢查對于病情的評估可能更為重要。Wang等[32]近期通過對5個家系6例ALMS患者進行分析,也證明了ERG和OCT對于ALMS診斷的重要性。另外,本研究中家系1先證者除典型視網膜血管、色素改變外,還出現沿血管少量點、線狀出血灶,考慮可能與其長期糖尿病且血糖控制不良相關。但在ALMS患者中,并不缺乏T2DM者,眼底出血卻少有報道,或許與ALMS患者的平均壽命有關,值得臨床進一步思考。
ALMS因早期缺乏全身表現,容易被誤診為Leber先天性黑矇、視網膜色素變性、全色盲、孤立性CORD等,這些遺傳性眼底疾病均可表現為早期的眼球震顫、畏光等[4]。Leber先天性黑矇患兒1歲以內即出現不追視,ERG顯示視錐視桿均為熄滅型;視網膜色素變性中夜盲為其特征,早期以視桿細胞損害為主,但ALMS患兒很少出現夜盲,可能是因為當視桿細胞受到損傷時患兒的視力已經非常差,反而感受不到暗視力的變化;全色盲是一種視錐細胞功能障礙性疾病,ERG通常顯示視錐細胞反應呈熄滅型,視桿細胞反應基本正常;ALMS通常比孤立性CORD患者經歷更嚴重且更早的視力損害[4]。當患者出現全身癥狀時,主要與Bardet-Biedl 綜合征(BBS)進行鑒別,BBS也是一種纖毛疾病,除CORD、肥胖、心肌病、腎功能異常、性功能減退、糖耐量異常外,還多出現多指或并指、智力低下等癥狀;另外,BBS患者的視力障礙出現在8歲左右,相對較ALMS更晚[13]。
CORD和嬰兒型擴張型心肌病是ALMS發生最早的臨床表現[11, 13]。我們總結的64例患者中以眼部表現為首發癥狀者占75.0%,這決定了眼科醫師對ALMS診斷的重要性。理想情況下發生眼球震顫的患兒均應接受全面眼科檢查,包括ERG和OCT,以及基因檢測,但因為家長對鎮靜的擔心和較高的基因檢測費用,使得一些遺傳性疾病無法得到早期診治。ALMS雖尚無有效治療方法,但早期診斷可進行心肌病的早期預防,學習盲文,訓練行動技能,通過日常飲食及運動改善肥胖,延緩T2DM發病時間等,可提高患者的生活質量。更多的治療ALMS的方法正在研究中[34]。
國內現有ALMS文獻中,均未詳細描述眼部表現。因該病臨床極為罕見,本研究僅納入我院確診的2個家系中的3例ALMS患者,并進行文獻復習,總結該病的眼部臨床特征,為臨床工作提供參考。目前仍缺乏長期隨訪的臨床資料,期待更多大樣本臨床研究對ALMS進行深入探討。