引用本文: 周鐘強, 魏圓夢, 唐賀, 彭海鷹, 史平玲, 李冠峰, 李苗. Alstr?m綜合征家系ALMS1基因變異分析. 中華眼底病雜志, 2023, 39(7): 538-543. doi: 10.3760/cma.j.cn511434-20230222-00136 復制
Alstr?m綜合征(ALMS)是一種由ALSM1基因變異所致并累及多系統的臨床罕見常染色體隱性遺傳病[1-3]。其臨床表現多樣,包括錐桿細胞營養不良、嚴重視力損傷、眼球震顫、兒童期肥胖、胰島素抵抗、2型糖尿病、感音神經性聽力損失、高甘油三酯血癥、擴張性心肌病、進行性肝腎功能不全、多器官纖維化等[1-3]。雖然ALMS是一種單基因疾病,但其致病的變異卻非常多,截止到2021年4月(此后未再更新),人類基因變異數據庫(HGMD)已收錄387個變異。此后又有數個新發變異被報道[4-10]。我們采用全外顯子測序對2個漢族ALMS家系進行檢測,在ALMS1基因發現2個新的致病變異。現將研究結果報道如下。
1 對象和方法
回顧性臨床研究。本研究通過河南省立眼科醫院倫理委員會審批[批文號:HNEECKY-2019(15號)];嚴格遵循《赫爾辛基宣言》原則;所有受試者及未成年監護人均獲知情并簽署書面知情同意書。
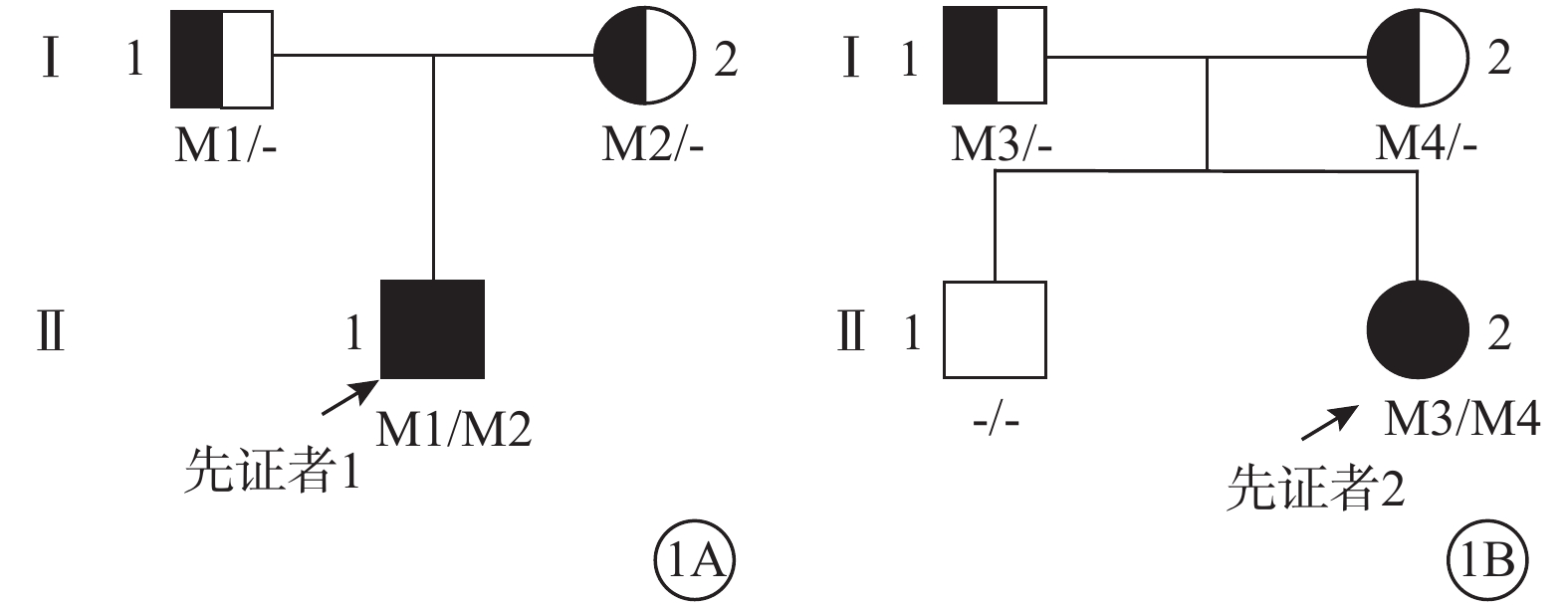
2020年8月至2021年12月于河南省立眼科醫院就診的2個ALMS家系的2例患者(先證者)及其家系成員5名納入本研究。患者分別來自2個無血緣關系家系(圖1),均為漢族。詳細詢問病史和家族史,并行最佳矯正視力(BCVA)、屈光度、裂隙燈顯微鏡聯合前置鏡、眼底彩色照相、色覺、頻域光相干斷層掃描(OCT)、全視野視網膜電圖(ff-ERG)檢查。患者均符合ALMS臨床診斷標準[11]。
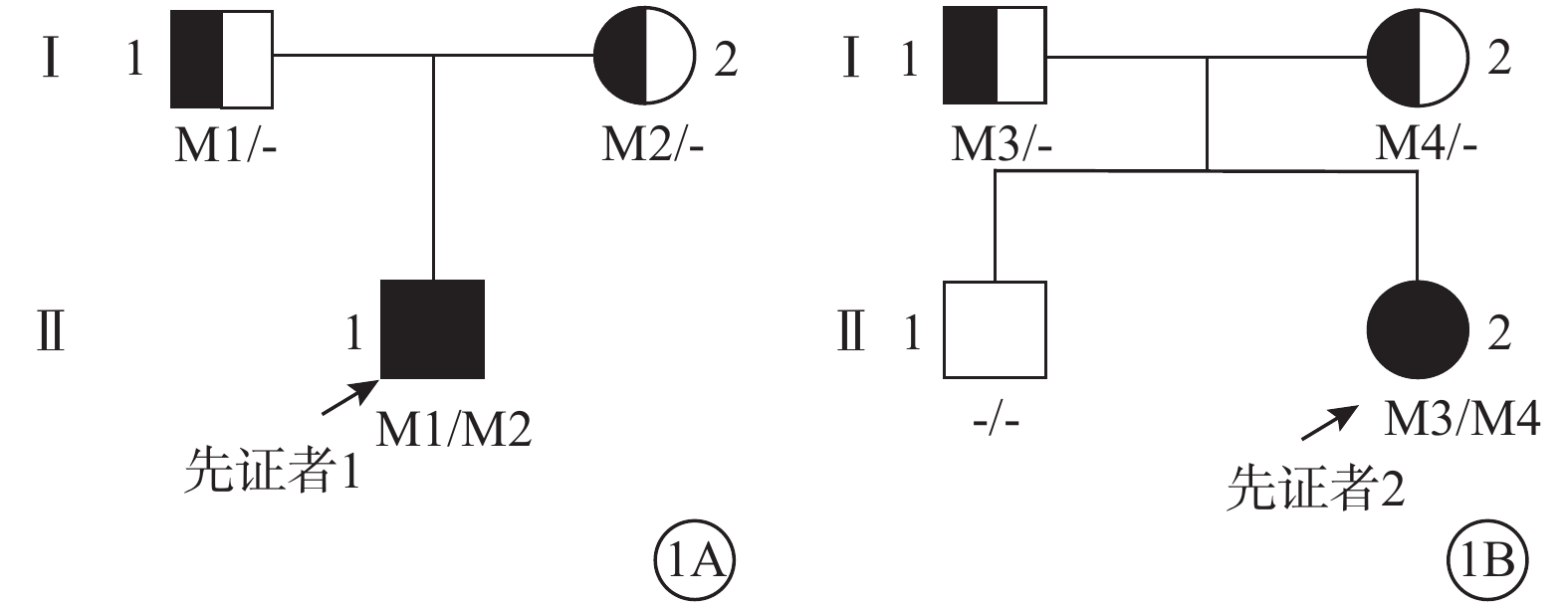 圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。□:正常男性;○:正常女性;↗:先證者;M1:c.468dupT(p.H156fs);M2:c.10819C>T(p.R3607X);M3:c.2296_2299del(p.Ser766Lysfs*13);M4:c.10831_10832del(p.Arg3611Alafs*6)
圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。□:正常男性;○:正常女性;↗:先證者;M1:c.468dupT(p.H156fs);M2:c.10819C>T(p.R3607X);M3:c.2296_2299del(p.Ser766Lysfs*13);M4:c.10831_10832del(p.Arg3611Alafs*6)
采用日本Canon公司CR-2 AF免散瞳眼底照相機和英國歐堡P200T激光掃描檢眼鏡行眼底照相;采用美國Optovue公司RTVue XR100-2 OCT儀和德國海德堡公司Spectralis OCT儀行OCT檢查;采用德國Roland公司RETI-Port 21眼電生理儀和法國Metrovision公司MonPack ONE眼電生理系統行ff-ERG檢查。
采集患者及其家系成員外周靜脈血5 ml,DNA提取試劑盒提取全基因組DNA。檢驗合格的DNA,通過Illumina HiSeq高通量測序平臺進行全外顯子測序,99%以上測序深度超過20×。測序結果應用Burrows-Wheeler Alignment Tool整理后與人基因組進行比對,使用GATK4消除假陽性、校正質量值并檢測和過濾單核苷酸多態性(SNP)和插入缺失標記結果。在外顯子組聚集聯盟(ExAC)數據庫(http://exac.broadinstitute.org/)、dbSNP數據庫(https://www.ncbi.nlm.nih.gov/snp/)、千人基因組計劃數據庫(http://www.1000genomes.org/)、人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)中查看變異在正常人群中的等位基因頻率,篩選出等位基因頻率小于1%的變異;根據ClinVar數據庫、HGMD及人類孟德爾遺傳在線數據庫(OMIM數據庫)查找變異位點的收錄情況。應用Mutation Taster軟件(http://www.mutationtaster.org/)預測氨基酸突變對蛋白功能的影響。參考OMIM、HGMD、ClinVar等數據庫對致病變異的異位點進行評估。對發現的致病變異位點進行Sanger測序檢驗。對數據的分析解讀參考美國醫學遺傳學和基因組學學院(ACMG)相關指南[12]。
2 結果
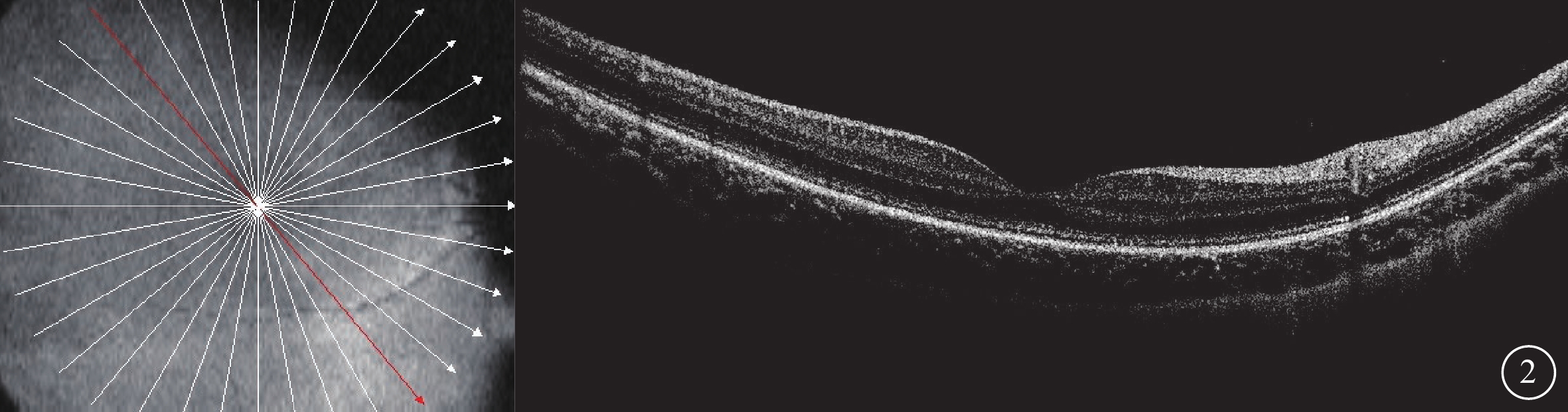
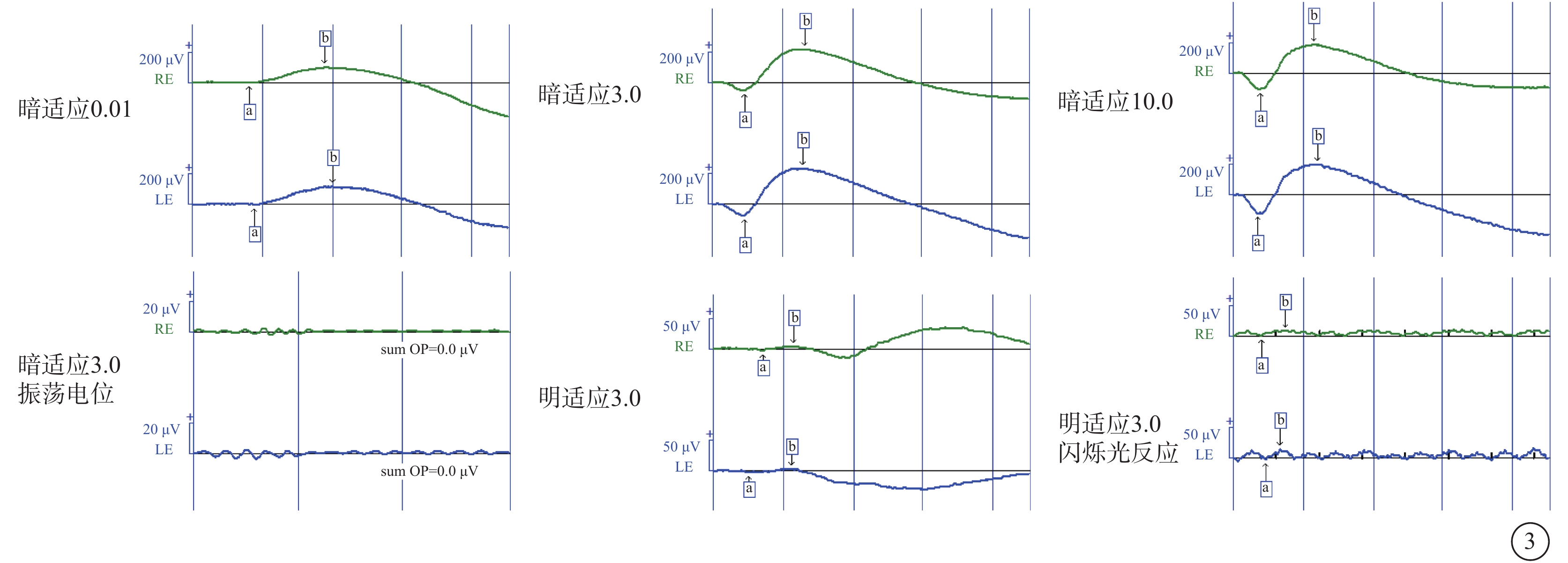
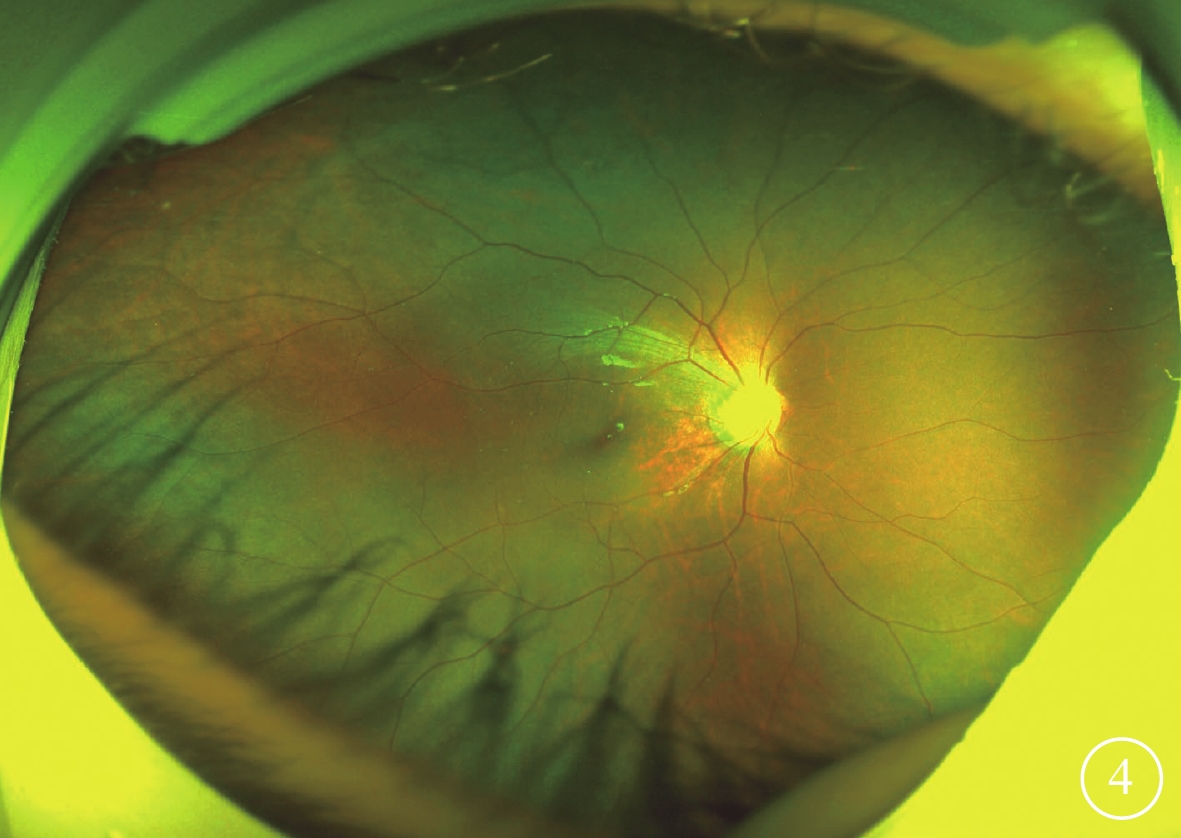
家系1先證者(Ⅱ1),男,初診年齡4歲7個月。雙眼自幼畏光伴視力低下,否認夜盲等癥狀。3月齡時因“心內膜彈力纖維增生癥、心功能不全、心肌受損、肝功能受損、左側腹股溝疝”于武漢某婦幼兒童專科醫院住院治療;4月齡時因“心內膜彈力纖維增生癥、重癥心肌炎?心功能不全、急性上呼吸道感染”于北京某兒童醫院住院治療。外院心臟超聲檢查,提示左室心內膜回聲增強,左室擴大并收縮功能減低,二尖瓣輕-中度關閉不全。初診眼科檢查,BCVA:右眼0.4(-2.75 DS/-2.50 DC×180°),左眼0.2(-2.00 DS/-2.75 DC×180°)。雙眼紅、綠色均不能辨認。雙眼均為中心凹注視;未見明顯眼球震顫。雙眼眼前節、眼底檢查未見明顯異常。OCT檢查,雙眼黃斑區橢圓體帶及嵌合體帶模糊、欠清晰(圖2)。ff-ERG檢查,雙眼視錐系統明顯受損,視桿系統輕度受損(圖3)。7歲1個月復診時眼科檢查,BCVA:右眼0.15(-4.00 DS/-2.50 DC×180°),左眼0.1(-4.50 DS/-3.50 DC×175°)。雙眼紅、綠色均不能辨認。雙眼均為中心凹注視;未見明顯眼球震顫。雙眼眼前節未見明顯異常。雙眼視網膜平伏,周邊大量色素沉著,黃斑中心凹反光不清(圖4)。掃頻源OCT檢查,雙眼黃斑區表現與初診時無明顯差異。先證者父親(Ⅰ1)、母親(Ⅰ2)眼部及全身檢查均未見明顯異常。
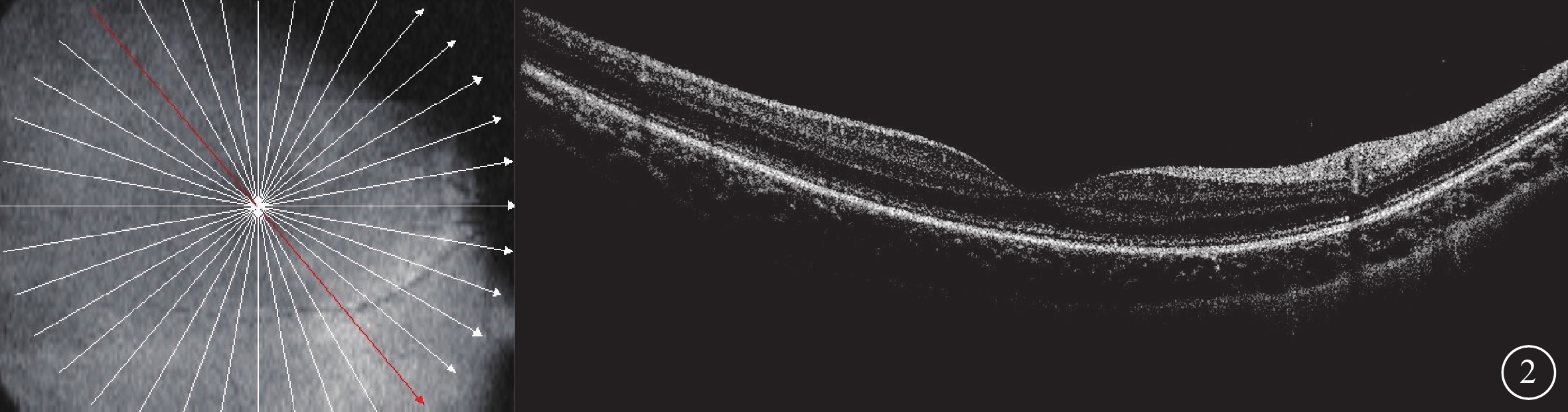 圖2
Alstr?m綜合征家系1先證者(Ⅱ1)初診右眼光相干斷層掃描像 左圖為掃描方向和部位,右圖為檢查結果。黃斑區橢圓體帶及嵌合體帶模糊、欠清晰
圖2
Alstr?m綜合征家系1先證者(Ⅱ1)初診右眼光相干斷層掃描像 左圖為掃描方向和部位,右圖為檢查結果。黃斑區橢圓體帶及嵌合體帶模糊、欠清晰
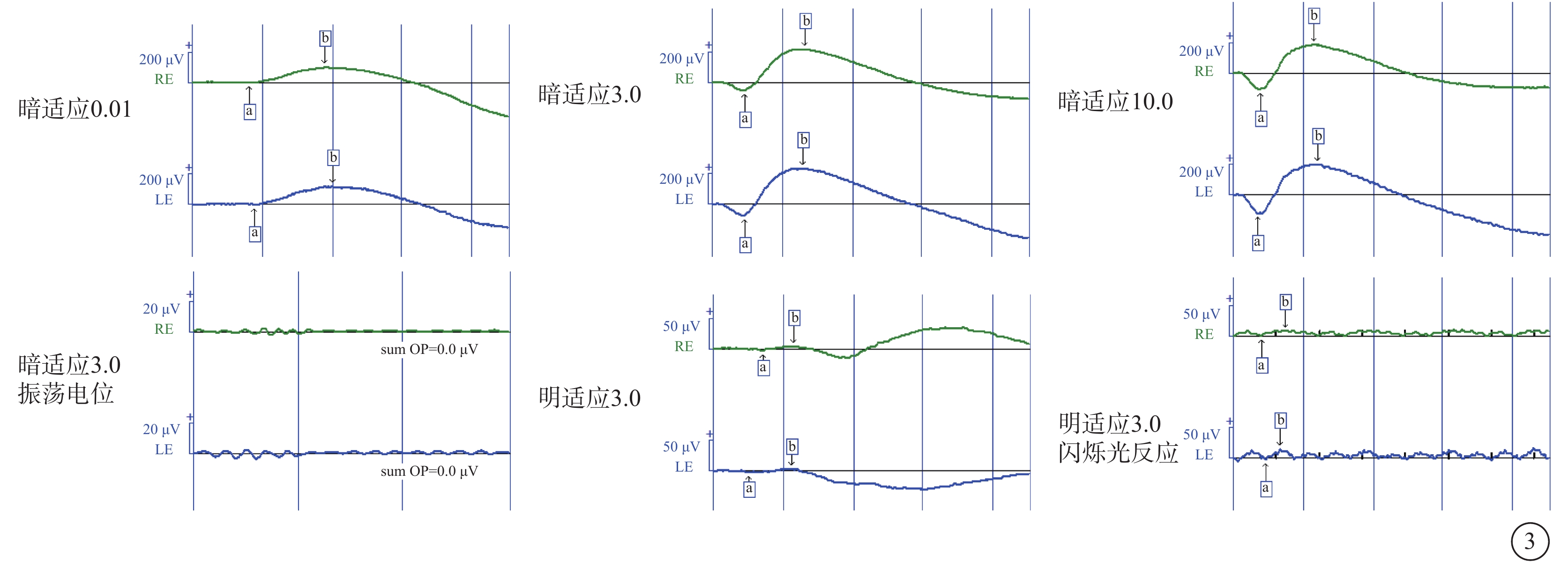 圖3
Alstr?m綜合征家系1先證者(Ⅱ1)初診時全視野視網膜電圖像 綠色波形為右眼,藍色波形為左眼。雙眼暗適應0.01 b波振幅輕度降低,3.0、10.0 a、b波振幅正常;暗適應振蕩電位無明確波形;明適應3.0 a、b波振幅嚴重降低,閃爍光反應振幅嚴重降低
圖3
Alstr?m綜合征家系1先證者(Ⅱ1)初診時全視野視網膜電圖像 綠色波形為右眼,藍色波形為左眼。雙眼暗適應0.01 b波振幅輕度降低,3.0、10.0 a、b波振幅正常;暗適應振蕩電位無明確波形;明適應3.0 a、b波振幅嚴重降低,閃爍光反應振幅嚴重降低
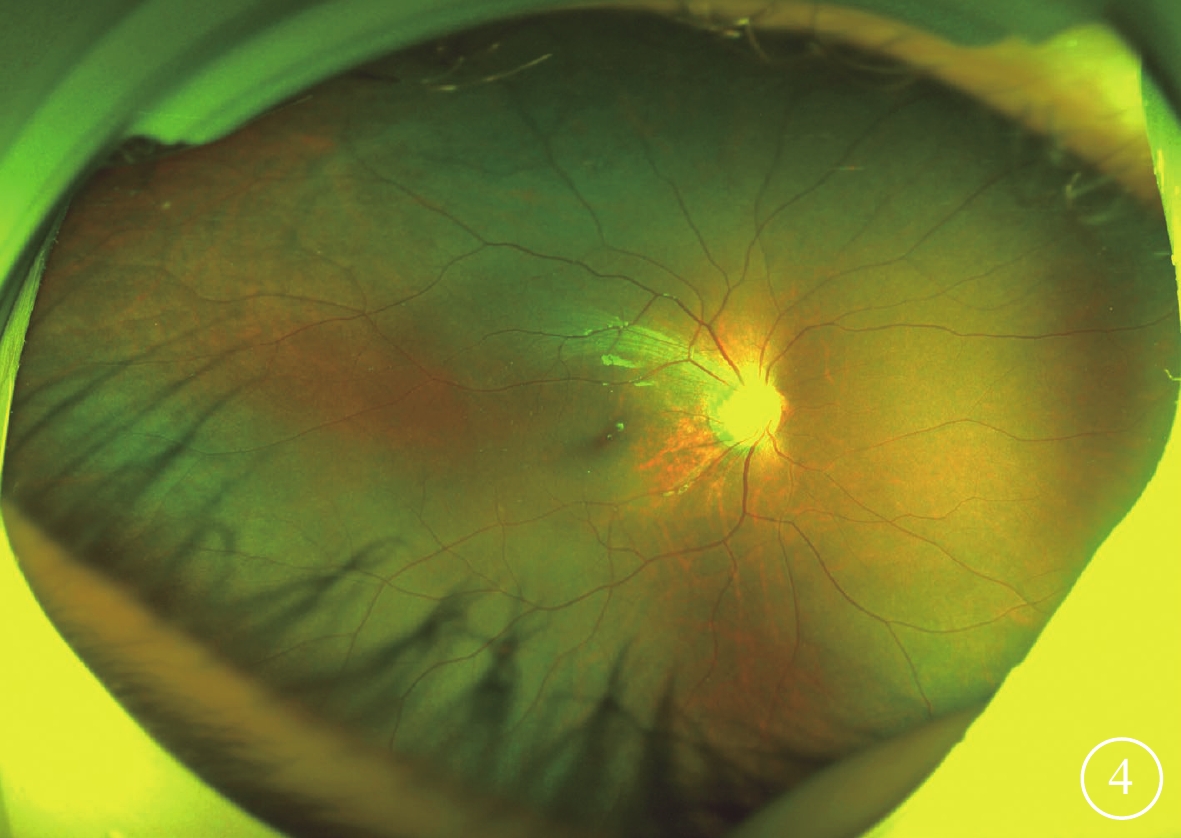 圖4
Alstr?m綜合征家系1先證者(Ⅱ1)復診時右眼彩色眼底像 視盤顏色正常、邊界清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,周邊視網膜色素沉著,黃斑中心凹反光不清
圖4
Alstr?m綜合征家系1先證者(Ⅱ1)復診時右眼彩色眼底像 視盤顏色正常、邊界清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,周邊視網膜色素沉著,黃斑中心凹反光不清
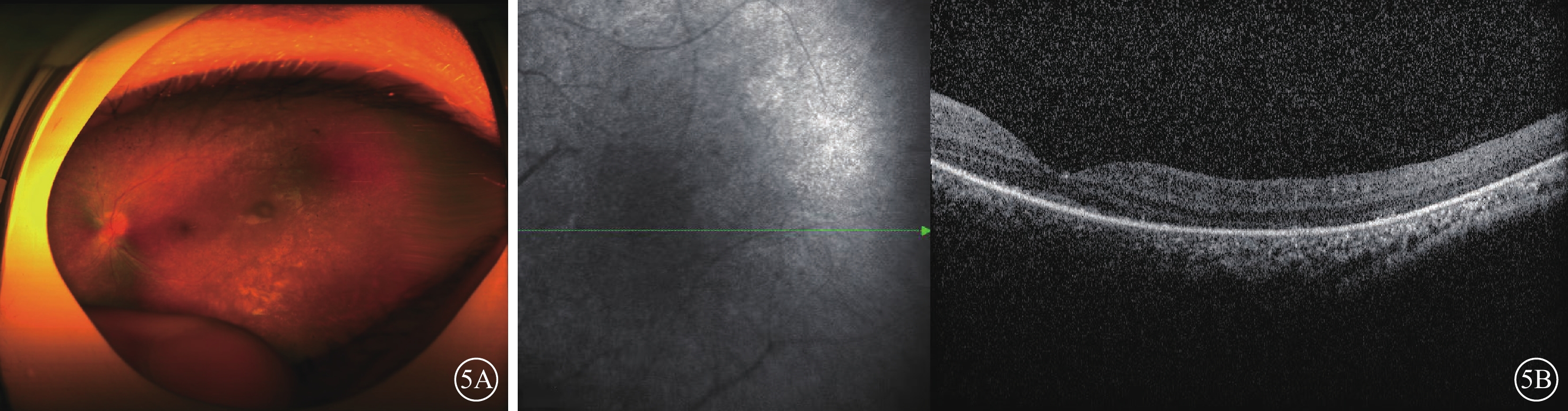
家系2先證者(Ⅱ2),女,12歲。雙眼自幼畏光伴視力低下。否認夜盲等癥狀。心臟彩色多普勒超聲檢查,提示心內大體結構及左心功能未見明顯異常;聽力檢查,提示雙耳高頻聽力損傷下降,存在感音神經性耳聾。眼科檢查,BCVA:右眼數指/1 m(+9.50 DS/-1.00 DC×75°),左眼手動/40 cm(+10.50 DS/-1.50 DC×180°)。雙眼紅、綠色均不能辨認。不能固視,明顯水平眼球震顫。雙眼眼前節未見明顯異常。雙眼視網膜平伏,大量色素沉著,黃斑中心凹反光不清(圖5A)。OCT檢查,雙眼視網膜前膜,神經視網膜外層結構紊亂并變薄,累及黃斑中心凹,視網膜色素上皮粗糙;右眼同時可見神經視網膜點狀中強反射信號,局部脈絡膜反射信號增強,伴光衰減(圖5B)。ff-ERG檢查,雙眼視錐視桿細胞嚴重受損(圖6A~6E)。先證者父親(Ⅰ1)、母親(Ⅰ2)、兄長(Ⅱ1)眼部及全身檢查均未見明顯異常。
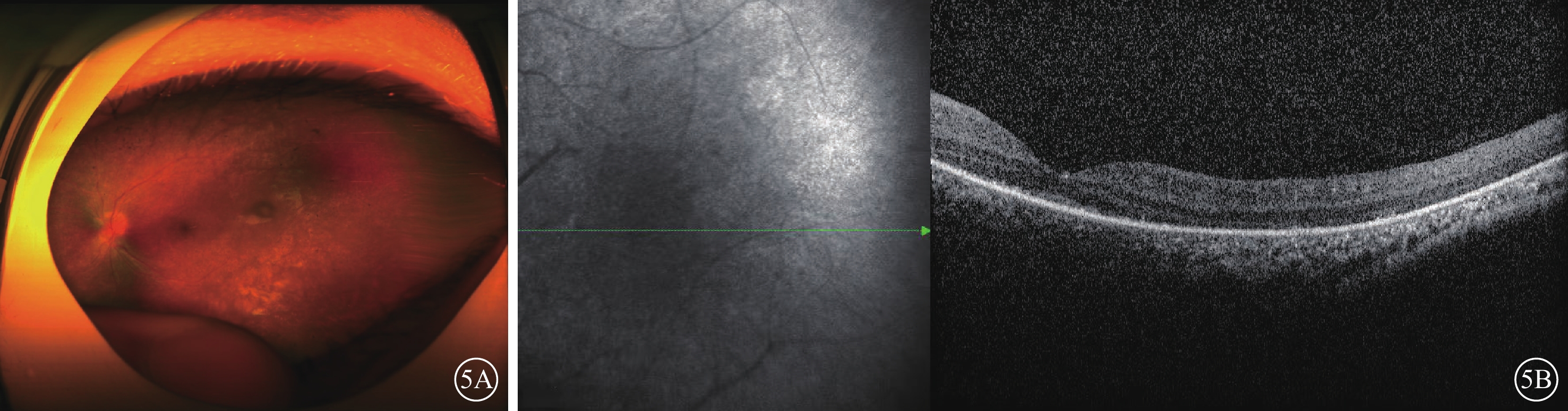 圖5
家系2先證者(Ⅱ2)左眼彩色眼底、光相干斷層掃描(OCT)像 5A示彩色眼底像,視盤顏色正常、邊界欠清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,大量色素沉著,黃斑中心凹反光不清。5B為OCT像,左圖為掃描方向和部位,右圖為檢查結果。視網膜前膜,神經視網膜外層結構紊亂并變薄,累及黃斑中心凹,視網膜色素上皮粗糙
圖5
家系2先證者(Ⅱ2)左眼彩色眼底、光相干斷層掃描(OCT)像 5A示彩色眼底像,視盤顏色正常、邊界欠清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,大量色素沉著,黃斑中心凹反光不清。5B為OCT像,左圖為掃描方向和部位,右圖為檢查結果。視網膜前膜,神經視網膜外層結構紊亂并變薄,累及黃斑中心凹,視網膜色素上皮粗糙
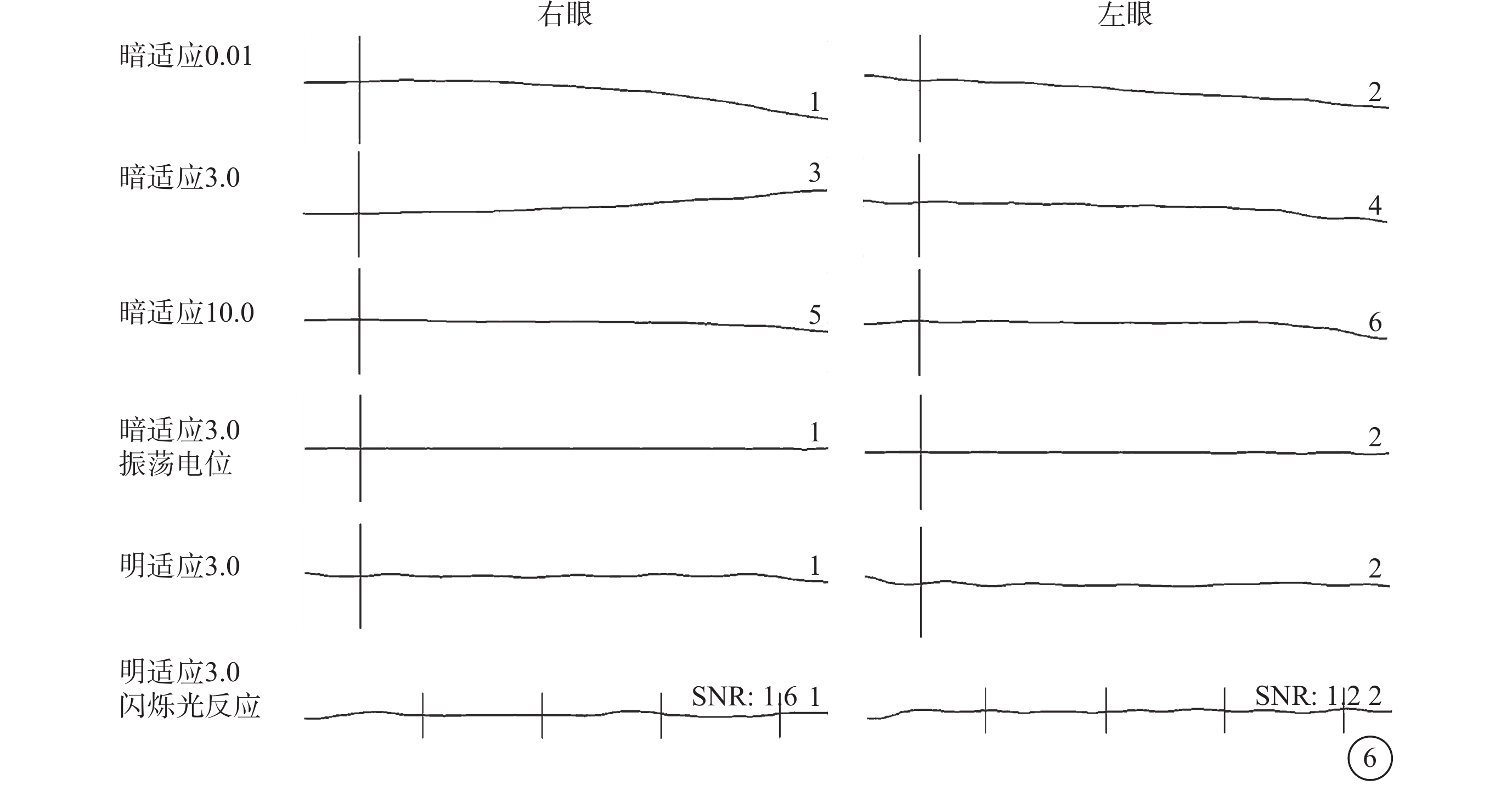 圖6
Alstr?m綜合征家系2先證者(Ⅱ2)全視野視網膜電圖像 暗適應0.01、3.0、10.0均無明確波形,暗適應3.0振蕩電位無明確波形;明適應3.0無明確波形;明適應3.0閃爍光反應無明確波形
圖6
Alstr?m綜合征家系2先證者(Ⅱ2)全視野視網膜電圖像 暗適應0.01、3.0、10.0均無明確波形,暗適應3.0振蕩電位無明確波形;明適應3.0無明確波形;明適應3.0閃爍光反應無明確波形
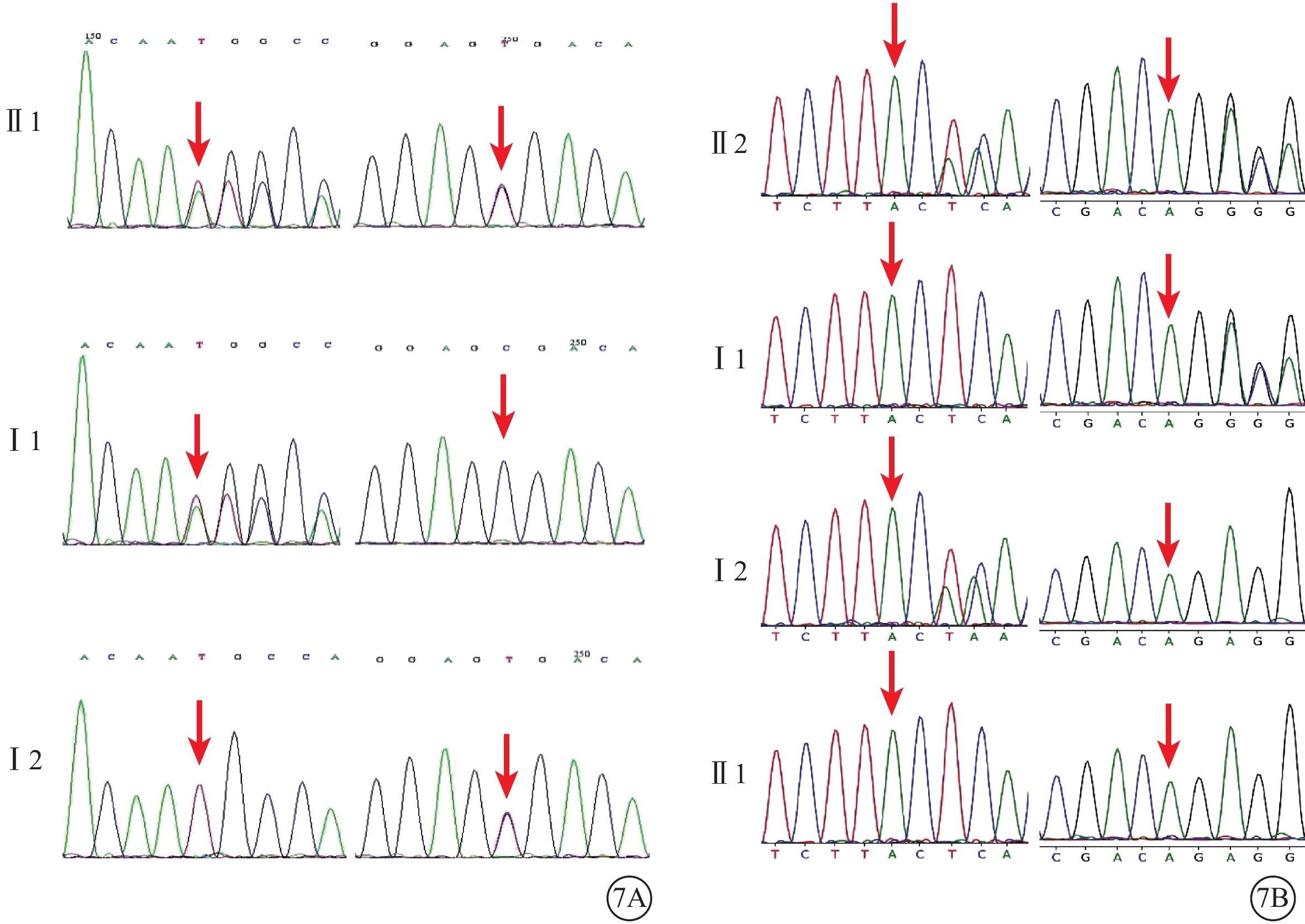
基因檢測結果顯示,家系1、2的ALMS1基因分別發現2個可能致病的變異和致病的變異(表1)。家系1的2個變異為c.468dupT(p.H156fs)(M1)、c.10819C>T(p.R3607X)(M2),分別為移碼變異和無義變異(PVS1),在ExAC等數據庫中均未見收錄(PM2),其中無義變異c.10819C>T通過核酸保守性預測為保守區域GERP++(5.66,Conserved)(PP3)。根據ACMG指南評估,c.468dupT為可能致病(PVS1+PM2),c.10819C>T為致病的無義變異(PVS1+PM2+PP3)。家系2的2個變異分別為c.2296_2299del(p.Ser766Lysfs*13)(M3)、c.10831_10832del(p.Arg3611Alafs*6)(M4),均為移碼變異(PVS1),在ExAC等數據庫中均未見收錄(PM2),其中M4經家系驗證屬于在反式(in trans)位置檢測到的致病性或疑似致病性變異(PM3)。根據ACMG指南評估,M3為可能致病(PVS1+PM2),M4為致病的移碼變異(PVS1+PM2+PM3)。2個家系中先證者均為復合雜合變異(圖7),其中家系1的2個變異為新發變異,既往未見文獻報道(表1)。
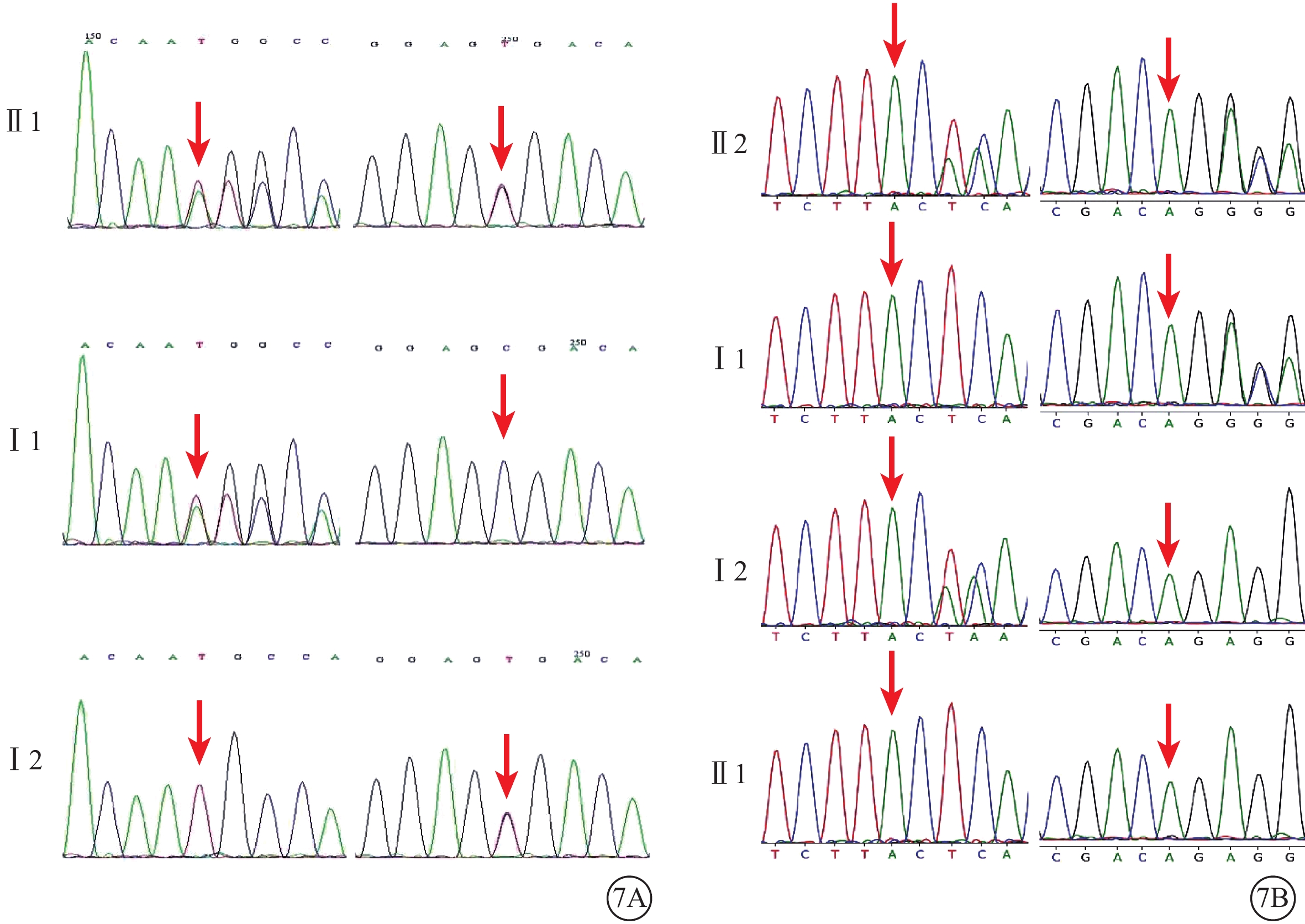 圖7
Alstr?m綜合征家系基因測序圖 7A示家系1,先證者(Ⅱ1)攜帶ALSM1基因c.468dupT(M1)移碼變異和c.10819C>T(M2)無義變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M1、M2變異(紅箭)。7B示家系2,先證者(Ⅱ2)攜帶ALSM1基因c.2296_2299del(M3)、c.10831_10832del(M4)移碼變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M3、M4變異(紅箭),其兄長(Ⅱ1)該位點不存在變異(紅箭)
圖7
Alstr?m綜合征家系基因測序圖 7A示家系1,先證者(Ⅱ1)攜帶ALSM1基因c.468dupT(M1)移碼變異和c.10819C>T(M2)無義變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M1、M2變異(紅箭)。7B示家系2,先證者(Ⅱ2)攜帶ALSM1基因c.2296_2299del(M3)、c.10831_10832del(M4)移碼變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M3、M4變異(紅箭),其兄長(Ⅱ1)該位點不存在變異(紅箭)
3 討論
本研究通過對2個ALMS家系的研究,確定家系1患者的ALMS1基因存在2個新發基因變異,即M1、M2。
本研究2例先證者均以畏光伴視力下降就診于我院門診。我們首先通過裂隙燈顯微鏡聯合前置鏡、驗光及眼位檢查將引起該癥狀的三大可能原因排除[13],進而將診斷范圍縮小到視錐系統本身病變的疾病。又根據患者既往的病史及相關系統輔助檢查(心臟超聲及聽力檢查)將錐桿細胞營養不良排除,進而將診斷鎖定為纖毛疾病。最終通過詳細眼部及全身病史采集、既往就診史、眼部及全身查體(全身查體未見多指、多趾等表現)及基因檢測結果,將患者診斷為ALMS。因此,對于畏光合并視力低下患者,我們建議除常規眼部病史詢問、眼部檢查及部分必要的輔助檢查外,應重視全身病史的采集及根據需要進行全身體格檢查。
ALMS1基因位于常染色體2p13.1,包含23個外顯子,編碼4 169個氨基酸組成的、相對分子質量為461.2×103的蛋白質[11]。有研究表明,ALMS1基因廣泛表達于細胞質、細胞骨架、微管組織中心、細胞中心體以及纖毛基底部;在細胞內物質運輸、細胞周期調控、纖毛細胞信號通路調節、維持纖毛細胞功能和結構穩定、細胞分化以及能量代謝平衡中起重要作用[14];對轉鐵蛋白內質體的運輸、葡萄糖轉運蛋白4型的運輸以及脂肪形成等生物過程均有影響[15]。且ALMS1分子及其所介導的Notch信號通路在ALMS發生和發展的過程中發揮著非常關鍵的作用[14]。細胞敲除實驗表明,ALMS1基因缺失會導致Notch受體在次級內體的聚集[15]。Notch信號通路的過度激活會導致一系列的影響,包括延長新生心肌細胞的增生期和惡化肝臟中胰島素的選擇性抵抗,這些影響均與ALMS的發病機制相關[2, 16]。本研究檢測到在ALSM1基因的第3、8、16號外顯子發生的4個變異最終都造成蛋白質翻譯提前終止,不能形成完整的蛋白質產物,這將對蛋白質功能產生極大影響。雖然ACMG評級僅將同樣位于第16號外顯子的M2和M4評為“致病的變異”,但是M1和M3分別位于第3、8號外顯子,蛋白質翻譯終止較M2和M4更早,形成的蛋白質產物更不完整,因此我們認為M1和M3更有可能具有致病性,并將在后續研究中驗證這一推測。
本研究的優勢在于通過詳細臨床研究,發現了2個ALMS家系,臨床工作中,該類患者常被誤診為視錐細胞營養不良、單純錐桿細胞營養不良或全色盲。本研究發現了2個新的變異位點,擴大了已報道基因的變異譜,對更深入了解ALMS的發病機制提供了一定幫助。然而,本研究尚存在以下不足:(1)僅2個家系的2代家族成員的核心家系,因此研究的外推性欠佳;(2)僅核心家系成員進行了檢測,未對其他家系成員進行研究,因此不能確認上述新的變異位點分別來源于整個家系的具體家族成員。
經過幾十年的基因測序發展,越來越多的致病基因變異被鑒定出來,遺傳性眼病的診斷和治療得到了長足的發展。ALMS是一種累及眼及全身多系統的罕見病,輕癥患者容易被眼科和(或)內科醫生漏診及誤診,因此詳細的全身體格檢查、眼部檢查及基因檢查對患者的診斷、生活指導及后續的遺傳咨詢必不可少。雖然目前沒有有效的病因治療手段,但隨著基因治療的發展,在不久的將來,該病可能得到更好的治療。
Alstr?m綜合征(ALMS)是一種由ALSM1基因變異所致并累及多系統的臨床罕見常染色體隱性遺傳病[1-3]。其臨床表現多樣,包括錐桿細胞營養不良、嚴重視力損傷、眼球震顫、兒童期肥胖、胰島素抵抗、2型糖尿病、感音神經性聽力損失、高甘油三酯血癥、擴張性心肌病、進行性肝腎功能不全、多器官纖維化等[1-3]。雖然ALMS是一種單基因疾病,但其致病的變異卻非常多,截止到2021年4月(此后未再更新),人類基因變異數據庫(HGMD)已收錄387個變異。此后又有數個新發變異被報道[4-10]。我們采用全外顯子測序對2個漢族ALMS家系進行檢測,在ALMS1基因發現2個新的致病變異。現將研究結果報道如下。
1 對象和方法
回顧性臨床研究。本研究通過河南省立眼科醫院倫理委員會審批[批文號:HNEECKY-2019(15號)];嚴格遵循《赫爾辛基宣言》原則;所有受試者及未成年監護人均獲知情并簽署書面知情同意書。
2020年8月至2021年12月于河南省立眼科醫院就診的2個ALMS家系的2例患者(先證者)及其家系成員5名納入本研究。患者分別來自2個無血緣關系家系(圖1),均為漢族。詳細詢問病史和家族史,并行最佳矯正視力(BCVA)、屈光度、裂隙燈顯微鏡聯合前置鏡、眼底彩色照相、色覺、頻域光相干斷層掃描(OCT)、全視野視網膜電圖(ff-ERG)檢查。患者均符合ALMS臨床診斷標準[11]。
圖1
Alstr?m綜合征家系圖 1A、1B分別示家系1、2。□:正常男性;○:正常女性;↗:先證者;M1:c.468dupT(p.H156fs);M2:c.10819C>T(p.R3607X);M3:c.2296_2299del(p.Ser766Lysfs*13);M4:c.10831_10832del(p.Arg3611Alafs*6)
采用日本Canon公司CR-2 AF免散瞳眼底照相機和英國歐堡P200T激光掃描檢眼鏡行眼底照相;采用美國Optovue公司RTVue XR100-2 OCT儀和德國海德堡公司Spectralis OCT儀行OCT檢查;采用德國Roland公司RETI-Port 21眼電生理儀和法國Metrovision公司MonPack ONE眼電生理系統行ff-ERG檢查。
采集患者及其家系成員外周靜脈血5 ml,DNA提取試劑盒提取全基因組DNA。檢驗合格的DNA,通過Illumina HiSeq高通量測序平臺進行全外顯子測序,99%以上測序深度超過20×。測序結果應用Burrows-Wheeler Alignment Tool整理后與人基因組進行比對,使用GATK4消除假陽性、校正質量值并檢測和過濾單核苷酸多態性(SNP)和插入缺失標記結果。在外顯子組聚集聯盟(ExAC)數據庫(http://exac.broadinstitute.org/)、dbSNP數據庫(https://www.ncbi.nlm.nih.gov/snp/)、千人基因組計劃數據庫(http://www.1000genomes.org/)、人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)中查看變異在正常人群中的等位基因頻率,篩選出等位基因頻率小于1%的變異;根據ClinVar數據庫、HGMD及人類孟德爾遺傳在線數據庫(OMIM數據庫)查找變異位點的收錄情況。應用Mutation Taster軟件(http://www.mutationtaster.org/)預測氨基酸突變對蛋白功能的影響。參考OMIM、HGMD、ClinVar等數據庫對致病變異的異位點進行評估。對發現的致病變異位點進行Sanger測序檢驗。對數據的分析解讀參考美國醫學遺傳學和基因組學學院(ACMG)相關指南[12]。
2 結果
家系1先證者(Ⅱ1),男,初診年齡4歲7個月。雙眼自幼畏光伴視力低下,否認夜盲等癥狀。3月齡時因“心內膜彈力纖維增生癥、心功能不全、心肌受損、肝功能受損、左側腹股溝疝”于武漢某婦幼兒童專科醫院住院治療;4月齡時因“心內膜彈力纖維增生癥、重癥心肌炎?心功能不全、急性上呼吸道感染”于北京某兒童醫院住院治療。外院心臟超聲檢查,提示左室心內膜回聲增強,左室擴大并收縮功能減低,二尖瓣輕-中度關閉不全。初診眼科檢查,BCVA:右眼0.4(-2.75 DS/-2.50 DC×180°),左眼0.2(-2.00 DS/-2.75 DC×180°)。雙眼紅、綠色均不能辨認。雙眼均為中心凹注視;未見明顯眼球震顫。雙眼眼前節、眼底檢查未見明顯異常。OCT檢查,雙眼黃斑區橢圓體帶及嵌合體帶模糊、欠清晰(圖2)。ff-ERG檢查,雙眼視錐系統明顯受損,視桿系統輕度受損(圖3)。7歲1個月復診時眼科檢查,BCVA:右眼0.15(-4.00 DS/-2.50 DC×180°),左眼0.1(-4.50 DS/-3.50 DC×175°)。雙眼紅、綠色均不能辨認。雙眼均為中心凹注視;未見明顯眼球震顫。雙眼眼前節未見明顯異常。雙眼視網膜平伏,周邊大量色素沉著,黃斑中心凹反光不清(圖4)。掃頻源OCT檢查,雙眼黃斑區表現與初診時無明顯差異。先證者父親(Ⅰ1)、母親(Ⅰ2)眼部及全身檢查均未見明顯異常。
圖2
Alstr?m綜合征家系1先證者(Ⅱ1)初診右眼光相干斷層掃描像 左圖為掃描方向和部位,右圖為檢查結果。黃斑區橢圓體帶及嵌合體帶模糊、欠清晰
圖3
Alstr?m綜合征家系1先證者(Ⅱ1)初診時全視野視網膜電圖像 綠色波形為右眼,藍色波形為左眼。雙眼暗適應0.01 b波振幅輕度降低,3.0、10.0 a、b波振幅正常;暗適應振蕩電位無明確波形;明適應3.0 a、b波振幅嚴重降低,閃爍光反應振幅嚴重降低
圖4
Alstr?m綜合征家系1先證者(Ⅱ1)復診時右眼彩色眼底像 視盤顏色正常、邊界清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,周邊視網膜色素沉著,黃斑中心凹反光不清
家系2先證者(Ⅱ2),女,12歲。雙眼自幼畏光伴視力低下。否認夜盲等癥狀。心臟彩色多普勒超聲檢查,提示心內大體結構及左心功能未見明顯異常;聽力檢查,提示雙耳高頻聽力損傷下降,存在感音神經性耳聾。眼科檢查,BCVA:右眼數指/1 m(+9.50 DS/-1.00 DC×75°),左眼手動/40 cm(+10.50 DS/-1.50 DC×180°)。雙眼紅、綠色均不能辨認。不能固視,明顯水平眼球震顫。雙眼眼前節未見明顯異常。雙眼視網膜平伏,大量色素沉著,黃斑中心凹反光不清(圖5A)。OCT檢查,雙眼視網膜前膜,神經視網膜外層結構紊亂并變薄,累及黃斑中心凹,視網膜色素上皮粗糙;右眼同時可見神經視網膜點狀中強反射信號,局部脈絡膜反射信號增強,伴光衰減(圖5B)。ff-ERG檢查,雙眼視錐視桿細胞嚴重受損(圖6A~6E)。先證者父親(Ⅰ1)、母親(Ⅰ2)、兄長(Ⅱ1)眼部及全身檢查均未見明顯異常。
圖5
家系2先證者(Ⅱ2)左眼彩色眼底、光相干斷層掃描(OCT)像 5A示彩色眼底像,視盤顏色正常、邊界欠清楚,視網膜動脈未見明顯變細、靜脈未見明顯紆曲,視網膜平伏,大量色素沉著,黃斑中心凹反光不清。5B為OCT像,左圖為掃描方向和部位,右圖為檢查結果。視網膜前膜,神經視網膜外層結構紊亂并變薄,累及黃斑中心凹,視網膜色素上皮粗糙
圖6
Alstr?m綜合征家系2先證者(Ⅱ2)全視野視網膜電圖像 暗適應0.01、3.0、10.0均無明確波形,暗適應3.0振蕩電位無明確波形;明適應3.0無明確波形;明適應3.0閃爍光反應無明確波形
基因檢測結果顯示,家系1、2的ALMS1基因分別發現2個可能致病的變異和致病的變異(表1)。家系1的2個變異為c.468dupT(p.H156fs)(M1)、c.10819C>T(p.R3607X)(M2),分別為移碼變異和無義變異(PVS1),在ExAC等數據庫中均未見收錄(PM2),其中無義變異c.10819C>T通過核酸保守性預測為保守區域GERP++(5.66,Conserved)(PP3)。根據ACMG指南評估,c.468dupT為可能致病(PVS1+PM2),c.10819C>T為致病的無義變異(PVS1+PM2+PP3)。家系2的2個變異分別為c.2296_2299del(p.Ser766Lysfs*13)(M3)、c.10831_10832del(p.Arg3611Alafs*6)(M4),均為移碼變異(PVS1),在ExAC等數據庫中均未見收錄(PM2),其中M4經家系驗證屬于在反式(in trans)位置檢測到的致病性或疑似致病性變異(PM3)。根據ACMG指南評估,M3為可能致病(PVS1+PM2),M4為致病的移碼變異(PVS1+PM2+PM3)。2個家系中先證者均為復合雜合變異(圖7),其中家系1的2個變異為新發變異,既往未見文獻報道(表1)。
圖7
Alstr?m綜合征家系基因測序圖 7A示家系1,先證者(Ⅱ1)攜帶ALSM1基因c.468dupT(M1)移碼變異和c.10819C>T(M2)無義變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M1、M2變異(紅箭)。7B示家系2,先證者(Ⅱ2)攜帶ALSM1基因c.2296_2299del(M3)、c.10831_10832del(M4)移碼變異(紅箭);其父親(Ⅰ1)、母親(Ⅰ2)分別攜帶M3、M4變異(紅箭),其兄長(Ⅱ1)該位點不存在變異(紅箭)
3 討論
本研究通過對2個ALMS家系的研究,確定家系1患者的ALMS1基因存在2個新發基因變異,即M1、M2。
本研究2例先證者均以畏光伴視力下降就診于我院門診。我們首先通過裂隙燈顯微鏡聯合前置鏡、驗光及眼位檢查將引起該癥狀的三大可能原因排除[13],進而將診斷范圍縮小到視錐系統本身病變的疾病。又根據患者既往的病史及相關系統輔助檢查(心臟超聲及聽力檢查)將錐桿細胞營養不良排除,進而將診斷鎖定為纖毛疾病。最終通過詳細眼部及全身病史采集、既往就診史、眼部及全身查體(全身查體未見多指、多趾等表現)及基因檢測結果,將患者診斷為ALMS。因此,對于畏光合并視力低下患者,我們建議除常規眼部病史詢問、眼部檢查及部分必要的輔助檢查外,應重視全身病史的采集及根據需要進行全身體格檢查。
ALMS1基因位于常染色體2p13.1,包含23個外顯子,編碼4 169個氨基酸組成的、相對分子質量為461.2×103的蛋白質[11]。有研究表明,ALMS1基因廣泛表達于細胞質、細胞骨架、微管組織中心、細胞中心體以及纖毛基底部;在細胞內物質運輸、細胞周期調控、纖毛細胞信號通路調節、維持纖毛細胞功能和結構穩定、細胞分化以及能量代謝平衡中起重要作用[14];對轉鐵蛋白內質體的運輸、葡萄糖轉運蛋白4型的運輸以及脂肪形成等生物過程均有影響[15]。且ALMS1分子及其所介導的Notch信號通路在ALMS發生和發展的過程中發揮著非常關鍵的作用[14]。細胞敲除實驗表明,ALMS1基因缺失會導致Notch受體在次級內體的聚集[15]。Notch信號通路的過度激活會導致一系列的影響,包括延長新生心肌細胞的增生期和惡化肝臟中胰島素的選擇性抵抗,這些影響均與ALMS的發病機制相關[2, 16]。本研究檢測到在ALSM1基因的第3、8、16號外顯子發生的4個變異最終都造成蛋白質翻譯提前終止,不能形成完整的蛋白質產物,這將對蛋白質功能產生極大影響。雖然ACMG評級僅將同樣位于第16號外顯子的M2和M4評為“致病的變異”,但是M1和M3分別位于第3、8號外顯子,蛋白質翻譯終止較M2和M4更早,形成的蛋白質產物更不完整,因此我們認為M1和M3更有可能具有致病性,并將在后續研究中驗證這一推測。
本研究的優勢在于通過詳細臨床研究,發現了2個ALMS家系,臨床工作中,該類患者常被誤診為視錐細胞營養不良、單純錐桿細胞營養不良或全色盲。本研究發現了2個新的變異位點,擴大了已報道基因的變異譜,對更深入了解ALMS的發病機制提供了一定幫助。然而,本研究尚存在以下不足:(1)僅2個家系的2代家族成員的核心家系,因此研究的外推性欠佳;(2)僅核心家系成員進行了檢測,未對其他家系成員進行研究,因此不能確認上述新的變異位點分別來源于整個家系的具體家族成員。
經過幾十年的基因測序發展,越來越多的致病基因變異被鑒定出來,遺傳性眼病的診斷和治療得到了長足的發展。ALMS是一種累及眼及全身多系統的罕見病,輕癥患者容易被眼科和(或)內科醫生漏診及誤診,因此詳細的全身體格檢查、眼部檢查及基因檢查對患者的診斷、生活指導及后續的遺傳咨詢必不可少。雖然目前沒有有效的病因治療手段,但隨著基因治療的發展,在不久的將來,該病可能得到更好的治療。